

(1).jpg)


Решение Совета Евразийской экономической комиссии от 4 июля 2023 г. № 77 “О внесении изменений в Правила проведения исследований биологических лекарственных средств Евразийского экономического союза”
В соответствии со статьей 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, пунктом 88 приложения № 1 к Регламенту работы Евразийской экономической комиссии, утвержденному Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. № 98, Совет Евразийской экономической комиссии решил:
1. Внести в Правила проведения исследований биологических лекарственных средств Евразийского экономического союза, утвержденные Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 89, изменения согласно приложению.
2. Настоящее Решение вступает в силу по истечении 180 календарных дней с даты его официального опубликования.
Члены Совета Евразийской экономической комиссии:
От Республики Армения
М. Григорян
От Республики Беларусь
И. Петришенко
От Республики Казахстан
С. Жумангарин
От Кыргызской Республики
А. Касымалиев
От Российской Федерации
А. Оверчук
ПРИЛОЖЕНИЕ
к Решению Совета Евразийской
экономической комиссии
от 4 июля 2023 г. № 77
Изменения,
вносимые в Правила проведения исследований биологических лекарственных средств Евразийского экономического союза
1. В предложении втором абзаца второго пункта 2.3.2.1 главы 1 слова «или в или иных» заменить словами «или в иных».
2. В предложении первом абзаца седьмого подраздела 1.1 раздела 1 главы 5.4 слова «трех R» (сократить/оптимизировать/заменить «reduce/refine/replace»)» заменить словами «3R (замена, улучшение и сокращение (replacement, refinement, reduction)».
3. В предложении третьем абзаца первого подраздела 4.3 раздела 4 главы 15.2 слова «(замена, улучшение, сокращение) (replacement, refinement, reduction)» заменить словами «(замена, улучшение и сокращение (replacement, refinement, reduction))».
4. В предложении пятом абзаца первого подраздела 4.3 раздела 4 главы 15.3 слова «(reduce - refine - replace, сокращение - улучшение - замена)» заменить словами «(замена, улучшение и сокращение (replacement, refinement, reduction))».
5. Главу 15.6 изложить в следующей редакции:
«Глава 15.6. Доклинические и клинические исследования биоаналогичных (биоподобных) лекарственных препаратов на основе гепаринов низкой молекулярной массы
1. Вступление
Гепарин является высоко сульфированным и гетерогенным членом семейства гликозаминогликановых углеводов, состоящих из различных дисахаридных единиц. К наиболее распространенным дисахарам относятся: 2-О-сульфат  -L-идуроновой кислоты и 6-О-сульфат, N-сульфат
-L-идуроновой кислоты и 6-О-сульфат, N-сульфат  -D-глюкозамин и IdoA (2S)-G1cNS (6S). Эндогенный гепарин вырабатывается в гранулах тучных клеток (лаброцитов) и обладает самой высокой плотностью отрицательного заряда среди всех биологических молекул.
-D-глюкозамин и IdoA (2S)-G1cNS (6S). Эндогенный гепарин вырабатывается в гранулах тучных клеток (лаброцитов) и обладает самой высокой плотностью отрицательного заряда среди всех биологических молекул.
Гепарин угнетает образование нескольких сериновых протеаз системы свертывания крови посредством активации антитромбина. Основную роль в связывании гепарина с антитромбином играет пентасахаридная последовательность, содержащая З-О-сульфатный глюкозаминовый остаток. После связывания с антитромбином гепарин вызывает конформационное изменение его молекулы, что приводит к активации области, ответственной за ингибирование активированных факторов свертывания. Кроме того, гепарин выступает в роли катализатора, связывая ингибитор и активированные протеазы серина, такие как тромбин (факторы II и IIa), факторы IXa и XIa. Это свойство гепарина зависит главным образом от числа моносахаридов и положения сульфатных групп в молекуле гепарина.
Молекулы гепарина, содержащие менее 18 моносахаридов, не катализируют ингибирование тромбина, но угнетают действие фактора свертывания Ха. После того как сериновые протеазы начинают действовать на специфичную пептидную связь Arg-Ser (аргинин-серин) активного центра молекулы антитромбина, гепарин повышает скорость реакции между тромбином и антитромбином как минимум в тысячу раз, с образованием стабильного комплекса 1:1. Кроме того, вклад в антитромботическое действие гепарина вносят и другие механизмы, независимые от антитромбина. Подобные механизмы включают высвобождение эндотелием сосудов ингибитора пути тканевого фактора, значимого естественного ингибитора системы коагуляции, взаимодействие с кофактором II гепарина, ингибирование прокоагулянтных эффектов лейкоцитов, стимуляция фибринолиза, а также влияние на эндотелий сосудов (как опосредованное рецепторами, так и рецептор-независимое).
Низкомолекулярные гепарины получают из нефракционированного гепарина в процессе химической или ферментативной деполимеризации. В результате процесса деполимеризации, низкомолекулярные гепарины преимущественно состоят из молекул, содержащих менее 18 моносахаридов. Данное снижение размера молекул сопровождается потерей тромбин-ингибирующей активности и усилением ингибирования фактора Ха по сравнению с нефракционированным гепарином.
Все имеющиеся в настоящее время на мировом рынке низкомолекулярные гепарины получаются из слизистой оболочки кишечника свиней. Наблюдаемая гетерогенность состава низкомолекулярных гепаринов обусловлена природой нефракционированного гепарина, а также технологией процесса деполимеризации (химический или ферментный).
Имеющиеся на рынке оригинальные низкомолекулярные гепарины отличаются по своим фармакокинетическим и фармакодинамическим свойствам. Из-за сложности определения низкомолекулярных гепаринов в крови невозможно оценить фармакокинетические свойства препаратов на основе гепаринов. Однако оценить всасывание и элиминацию низкомолекулярных гепаринов можно с использованием фармакодинамических тестов, наиболее важными из которых является определение анти-Ха и анти-IIa активности.
Все оригинальные низкомолекулярные гепарины имеют терапевтические показания для лечения венозных тромбозов (лечение и профилактика венозной тромбоэмболии, а некоторые также имеют дополнительные показания, связанные с артериальным тромбозом и острым коронарным синдромом и инфарктом миокарда.
Наиболее частой нежелательной реакцией при применении гепаринов является кровотечение, а к наиболее серьезной нежелательной реакции относится редко наблюдаемая гепарин-индуцированная тромбоцитопения II типа, которая развивается под влиянием образования антител к новым антигенам, которые образуются при формировании комплекса, содержащего гепарин и тромбоцитарный фактор 4 (HPF4). Связывание антител с новым антигеном (комплекс гепарина и тромбоцитарного фактора 4 (HPF4)) вызывает активацию тромбоцитов с последующим образованием тромбогенных микроагрегатов тромбоцитов. У пациентов с иммуно-модулированной гепарин-индуцированной тромбоцитопенией повышен риск артериальных и венозных тромбоэмболических осложнений (гепарин-индуцированная тромбоцитопения и тромбоз). Хотя, по сравнению с нефракционированным гепарином, частота появления антител к комплексу гепарина и тромбоцитарного фактора 4 (HPF4) и развития гепарин-индуцированной тромбоцитопении II типа является более низкой на фоне применения нефракционированного гепарина, при назначении препаратов низкомолекулярного гепарина необходимо регулярно контролировать содержание тромбоцитов у всех пациентов, а у тех пациентов, у которых выявлена тромбоцитопения или тромбоэмболические осложнения, необходимо определение антител к комплексу, содержащему гепарин и тромбоцитарный фактор 4 (HPF4).
Настоящая глава применяется в дополнение к требованиям для установления подобия (сходства) двух препаратов в соответствии с главой 15 настоящих Правил. Требования настоящей главы необходимо рассматривать совместно с другими соответствующими требованиями и актами, органов Союза в сфере обращения лекарственных средств.
2. Сфера применения
Настоящая препарат-специфичная глава содержит доклинические и клинические требования к подтверждению биоаналогичности (биоподобия) двух лекарственных препаратов, содержащих низкомолекулярный гепарин, при этом необходимо учитывать следующие специфические аспекты качества в рамках сравнительной разработки:
1) должна быть доступна информация о биологическом источнике биоаналогичного (биоподобного) низкомолекулярного гепарина, процессе производства нефракционированного гепарина, режиме деполимеризации и соответствующих условий для данного процесса. Биоаналогичный (биоподобный) и оригинальный (референтный) низкомолекулярные гепарины необходимо детально охарактеризовать и сравнить, используя современные методы. Соответствие требованиям Фармакопеи Союза является минимальным стандартным требованием;
2) сравнительный анализ физико-химических и биологических параметров биоаналогичного (биоподобного) и оригинального (референтного) низкомолекулярного гепарина должен показать высокую степень сопоставимости в отношении:
распределения молекулярного веса и общего химического состава; исходного материала (тип ткани и биологический вид) и режима деполимеризации;
строительных блоков дисахаридов, профилей сопоставления фрагментов и последовательностей избранных нефрагментированных олигосахаридов;
биологических и биохимических методов анализа.
3. Связь с другими главами
В главах 15 - 15.2 настоящих Правил содержатся общие указания по разработке биоаналогичных (биоподобных) лекарственных препаратов.
4. Доклинические исследования
Перед началом клинических исследований должны быть проведены доклинические исследования. Доклинические исследования носят сравнительный характер и направлены на выявление различий между действием биоаналогичного (биоподобного) лекарственного препарата и оригинального (референтного) низкомолекулярного гепарина, а не на изучение фармакологического (клинического) ответа как такового на введение лекарственного препарата. Выбор подхода должен быть полностью обоснован в обзоре доклинических данных.
Исследование фармакодинамики
Исследование in vitro
Для сопоставления фармакодинамической активности подобного лекарственного препарата и оригинального (референтного) низкомолекулярного гепарина должны быть представлены данные нескольких сравнительных биологических анализов (на основании современных данных о клинически значимом фармакодинамическом влиянии низкомолекулярных гепаринов, включая как минимум оценку анти-Ха и анти-IIa активности). Для измерения активности необходимо использовать стандартизированные методы (например, в соответствии с Фармакопеей Союза). Такие данные могут быть получены ранее в процессе изучения качества лекарственного препарата.
Исследование in vivo
Если при характеристике физико-химических и биологических свойств с использованием высокочувствительных современных методов установлена высокая степень сходства биоаналогичного (биоподобного) и оригинального (референтного) препаратов, проведение исследований in vivo как части изучения сопоставимости не требуется. В остальных случаях in vivo сравнительное количественное изучение фармакодинамической активности биоаналогичного (биоподобного) и оригинального (референтного) низкомолекулярных гепаринов включает в себя:
использование фармакодинамической модели in vivo, разработанной с учетом самых современных данных о клинически значимых фармакодипамических эффектах низкомолекулярных гепаринов, в которую включена по меньшей мере оценка анти-Ха и анти-IIa активности, а также оценка степени высвобождения ингибитора пути тканевого фактора;
использование подходящей модели венозного, либо артериального тромбоза на животных, в соответствии с клиническими показаниями.
Исследование токсичности
Как правило, изучение токсичности при повторных введениях одной дозы не требуется.
В определенных случаях (например, если в состав препарата включено новое или малоизученное вспомогательное вещество), необходимо проведение дополнительных исследований токсичности.
Проведение сравнительных исследований неспецифической токсичности только для оценки установленных различий в составе примесей не следует проводить. Наилучшей стратегией для снижения рисков, обусловленных примесями (например, белками) является сведение содержания примесей к минимуму в соответствии с требованиями фармакопейной статьи (монографии) Фармакопеи Союза, а при отсутствии в ней - с требованиями фармакопей государств-членов.
В случае если иммуногенность не оценивается в рамках клинического исследования, иммуногенный потенциал биоаналогичного (биоподобного) и оригинального (референтного) низкомолекулярного гепарина необходимо сравнить в соответствующих доклинических исследованиях. Предсказательная ценность исследований на животных в отношении иммуногенности у человека обычно считается низкой. Однако для того, чтобы охарактеризовать физико-химические свойства комплексов гепарина и тромбоцитарного фактора 4 (HPF4), допускается проводить исследования in vitro. Современные методы анализа позволяют определить связывающую афинность низкомолекулярного гепарина к тромбоцитарному фактору 4, стехиометрии, заряд и размер итоговых комплексов и изменения в частоте появления вторичных структурных элементов (альфа-спиралей и бета-листов) в белке тромбоцитарного фактора 4. В этой связи данные характеристики комплекса гепарин - тромбоцитарный фактор 4 должны быть определены как функция концентрации и отношения содержания низкомолекулярного гепарина и тромбоцитарного фактора 4. Кроме того, необходимо рассмотреть возможность изучения способности комплексов гепарина и тромбоцитарного фактора 4 (HPF4) связываться с ранее сформировавшимися антителами против комплексов гепарина и тромбоцитарного фактора 4 (HPF4) и активировать тромбоциты, с использованием сыворотки от пациентов с гепарин-индуцированной тромбоцитопенией II типа.
Целесообразность любого использованного метода должна быть соответствующим образом обоснована. Для того чтобы продемонстрировать достаточную чувствительность соответствующих методов, в качестве положительного контроля допускается использовать нефракционированный гепарин (который обладает большей иммуногенностью по сравнению с низкомолекулярными гепаринами). Исследования фармакологической безопасности, репродуктивной токсичности не являются обязательными при сравнительном исследовании биоаналогичного (биоподобного) и оригинального (референтного) низкомолекулярных гепаринов. Исследования местной переносимости не проводятся в случае, если в состав препарата не включены вспомогательные вещества, для которых не имеется достаточного документально подтвержденного опыта использования при данном пути введения лекарственного препарата или же это опыт использования ограничен. Если выполнялись иные исследования in vivo, то оценку местной переносимости допускается проводить как часть таких исследований.
5. Клинические исследования
Исследование фармакокинетики и фармакодинамики
Гетерогенность низкомолекулярных гепаринов не позволяет проводить обычное исследование фармакокинетических свойств. Вместо этого должно быть проведено сравнение фармакодинамической активности, прежде всего в отношении анти-Ха и анти-IIa, между биоаналогичным (биоподобным) и оригинальным (референтным) низкомолекулярным гепарином. Кроме того, необходимо сравнить соотношение активности анти-Ха и анти-IIa факторов, равно как и активность ингибитора пути тканевого фактора. Изучение подобия (сходства) биоаналогичного (биоподобного) и оригинального (референтного) препаратов по фармакодинамическим показателям проводят в рандомизированном перекрестном и желательно двойном слепом исследовании в двух группах здоровых добровольцев при подкожном введении препаратов. Поскольку подкожное введение лекарственного препарата позволяет охарактеризовать как абсорбцию, так и элиминацию низкомолекулярного гепарина, дополнительные фармакологические исследования для внутривенного или внутриартериального применения (если применимо), не требуются.
Выбранная доза должна соответствовать чувствительной (крутой) части кривой зависимости «доза - эффект». Пределы эквивалентности также должны быть определены и должным образом обоснованы заранее.
Исследование эффективности
Ключевые доказательства сопоставимой эффективности должны быть получены на основании подобия, продемонстрированного в физико-химических, функциональных и фармакологических сравнительных исследованиях. Проведение отдельного клинического исследования сравнительной эффективности не требуется.
Исследование безопасности
Биоаналогичный (биоподобный) и оригинальный (референтный) низкомолекулярные гепарины должны продемонстрировать убедительное подобие физико-химических и функциональных характеристик и фармакодинамических профилей. В таком случае ожидается, что побочные эффекты, связанные с избыточно выраженными фармакологическими эффектами (например, кровотечение), будут наблюдаться со сходными частотами. Если помимо этого профиль примесей и природа вспомогательных веществ биоаналогичного (биоподобного) препарата не вызывает неопределенности в отношении их влияния на безопасность и (или) иммуногенность, исследование безопасности и (или) иммуногенности допускается не проводить. В этом случае, необходимо провести дальнейшее изучение иммуногенного потенциала в рамках доклинических исследований, как описано в разделе 4 настоящей главы.
В противном случае сравнительные данные по безопасности и (или) иммуногенности у пациентов должны быть получены до регистрации.
В таком клиническом исследовании, оценка иммуногенности должна включать в себя определение антител к комплексу гепарина и тромбоцитарного фактора 4 и обязательный мониторинг числа тромбоцитов с целью выявления случаев гепарин-индуцированной тромбоцитопении II типа. Кроме того, обширные и клинически значимые необширные кровотечения должны быть зарегистрированы и описаны. При этом необходимо использовать согласованную и клинически релевантную классификацию кровотечений. Оптимальным подходом является подход, при котором решение о геморрагических явлениях выносится центральным комитетом независимых экспертов на основании ослепленных данных.
6. План фармаконадзора
При регистрации биоаналогичного (биоподобного) препарата необходимо представить план управления рисками в соответствии с Правилами практики фармаконадзора. В плане управления рисками должны быть отражены выявленные и потенциальные риски применения оригинального (референтного) препарата в соответствии с инструкцией по применению оригинального (референтного) препарата, а также мероприятия по отслеживанию параметров безопасности применения для соответствующих показаний оригинального (референтного) препарата, на которые проводилась экстраполяция результатов исследований по другим показаниям. Кроме того, необходимо представить план управления рисками, связанными с серьезными нежелательными явлениями при приеме препаратов низкомолекулярных гепаринов (например, гепарин-индуцированная тромбоцитопения II типа, анафилактические и анафилактоидные реакции).
7. Экстраполяция показаний
Подтверждение биоподобия, основанного на физико-химических и функциональных характеристиках, фармакодинамических профилях и, данных исследования безопасности и (или) иммуногенности (где необходимо), позволяет экстраполировать результаты на другие пути введения и показания к применению, зарегистрированные у оригинального (референтного) препарата, если применимо и должным образом обосновано.».
6. Дополнить главами 24 - 30 следующего содержания:
«Глава 24. Указания по оценке производственного процесса препаратов из плазмы крови человека в отношении риска прионовой инфекции
1. Общие положения
1. Вариантная форма болезни Крейцтфельда-Якоба (вБКЯ) впервые была зарегистрирована в 1996 году. Достоверно установлено, что вариантную болезнь Крейцтфельда-Якоба вызывает передающийся человеку возбудитель губчатой энцефалопатии крупного рогатого скота. Наиболее вероятной причиной заболевания считают употребление в пищу продуктов, контаминированных трансмиссивными агентами губчатой энцефалопатии крупного рогатого скота.
2. В настоящее время заболеваемость вариантной болезнью Крейцтфельда-Якоба не поддается прогнозированию. В отличие от спорадической форы вариантная форма болезни Крейцтфельда-Якоба, характеризуется обширным вовлечением в патологический процесс лимфоретикулярной системы, что вызывает вероятность передачи заболевания через кровь или лекарственные препараты, полученные из крови человека, находящегося в инкубационном периоде заболевания. Прионная опасность компонентов крови подтверждена при проведении экспериментальных исследований на грызунах. Также была обнаружена инфекционность лейкоцитарной пленки серого мышиного лемура (Microcebus murinus), которого в лабораторных условиях инфицировали штаммом губчатой энцефалопатии, адаптированным к макакам.
3. При проведении межвидовых гемотрансфузий установлено, что как заболевание губчатая энцефалопатия, вызванная в условиях эксперимента так и природная инфекция скрейпи могут передаваться при переливания крови у овец. В других исследованиях показано, что переливание крови человека диким и трансгенным мышам и обезьянам не вызывает заболевание, но исследования еще не завершены. В Великобритании зарегистрированы 2 случая заболевания вариантной болезнью Крейцтфельда-Якоба, возможной причиной которых рассматривают переливание эритроцитов доноров, у которых ретроспективно обнаружены симптомы вариантной болезни Крейцтфельда-Якоба. На сегодняшний день не зарегистрировано ни одного случая заболевания этой болезнью, связанного с введением препаратов крови (исследования проводились в группах реципиентов высокого риска, таких как больные гемофилией). Накопленных эпидемиологических данных о вариантной болезни Крейцтфельда- Якоба недостаточно для оценки риска передачи трансмиссивных агентов губчатой энцефалопатии при применении препаратов крови. В качестве меры предосторожности плазма доноров из Великобритании в настоящее время не используется для производства препаратов крови, поскольку в Великобритании было зарегистрировано наибольшее количество случаев губчатой энцефалопатии и значительно большее, чем в любой иной стране, количество случаев вариантной болезни Крейцтфельда-Якоба.
2. Сфера применения
4. Современные технологии производства препаратов крови обеспечивают значительное снижение инфекционности агентов, возможно присутствующих в плазме крови человека.
5. Производители должны оценивать вклад стадий производственного процесса в снижение инфекционности с использованием взаимодополняющих методов оценки.
6. Настоящая глава содержит указания по оценке процесса производства препаратов крови в отношении риска передачи вариантной болезни Крейцтфельда-Якоба.
3. Исследования по оценке процедур очистки от трансмиссивных агентов губчатой энцефалопатии
3.1. Общие принципы
7. Указания, приведенные в главе 3 настоящих Правил распространяются и на трансмиссивные агенты губчатой энцефалопатии (ТАГЭ).
8. В промежуточные продукты добавляется материал с содержанием не более 10% определенного трансмиссивного агента. Исследования по оценке процедур очистки необходимо проводить в условиях уменьшенного масштаба процесса производства, моделирующего реальный производственный процесс. Исследования должны проводиться персоналом, имеющим надлежащую квалификацию в специально оборудованной лаборатории с тщательным документированием всех процедур.
9. Необходимо доказать валидность (пригодность) уменьшенного масштаба производства и подтвердить максимальное соответствие уровня очистки в уменьшенном масштабе процесса производства полномасштабному процессу промышленного производства.
10. Исследованию подлежат только стадии производства, которые считаются эффективными для инактивации и (или) удаления трансмиссивных агентов губчатой энцефалопатии.
11. Трансмиссивные агенты губчатой энцефалопатии устойчивы к большинству физико-химических процедур инактивации, традиционно используемых в процессе производства препаратов крови. В связи с этим особое внимание необходимо уделять таким стадиям удаления (разделения), как фракционирование этанолом при низких температурах, осаждение полиэтиленгликолем, хроматография, глубинная фильтрация или нанофильтрация. Валидационные исследования указанных стадий необходимо проводить не только в отношении вирусов, но и для оценки удаления трансмиссивных агентов губчатой энцефалопатии.
12. Исследования процедур очистки от трансмиссивных агентов губчатой энцефалопатии более трудоемкие и дорогостоящие, по сравнению с исследованиями традиционных вирусов, поэтому допускается проведение теоретического анализа данных, подтверждающих робастность процесса производства или использование валидированных методов in vitro.
13. Изменения параметров производственного процесса оказывают влияние на удаление трансмиссивных агентов губчатой энцефалопатии. Поэтому их необходимо учитывать при разработке дизайна валидационных исследований стадий разделения.
14. Валидационные исследования должны включать в себя оценку методами in vitro разделения прионных частиц в промежуточных фракциях. Если коэффициент снижения одной стадии составляет 1,0 lg или менее, то он признается незначительным, и не учитывается в расчете суммарного фактора (коэффициента) снижения. Титр трансмиссивного агента губчатой энцефалопатии в материале, добавляемом при проведении валидационных исследований, может оказаться достаточно высоким для оценки вклада двух и более стадий. Поэтому дизайн валидационных исследований должен позволять оценить коэффициенты снижения для каждой стадии отдельно и в комбинации при оценке промежуточных продуктов. Полученные коэффициенты снижения для каждой стадии суммируются для обоснования общего вклада всех стадий в процесс удаления трансмиссивных агентов губчатой энцефалопатии, включая стадии, которые внесли незначительный вклад.
15. Валидационные исследования нескольких стадий пригодны и в случае возможного изменения физико-химических свойств трансмиссивного агента губчатой энцефалопатии на определенном этапе производства, которые могут оказать влияние на эффективность следующей стадии удаления (например, при обработке детергентом перед этапом фильтрации).
16. Необходимо стремиться к проведению валидационных исследований всех стадий процесса производства по оценке способности удалять трансмиссивные агенты губчатой энцефалопатии.
17. Однако существуют ограничения исследований, связанные с недостаточным содержанием (титром) трансмиссивного агента губчатой энцефалопатии в добавляемом материале для оценки снижения в ходе двух и более стадий производственного процесса.
18. Основными факторами, требующими внимания являются:
уменьшенный масштаб производства (модельный);
выбор добавляемого трансмиссивного агента губчатой энцефалопатии;
выбор метода оценки количественного содержания добавляемого трансмиссивного агента губчатой энцефалопатии; выбор стадий производственного процесса; интерпретация данных и ограничения исследований; повторные исследования по оценке процедур очистки от трансмиссивных агентов губчатой энцефалопатии;
санитарная очистка промышленного оборудования.
3.2. Уменьшенный масштаб производства (модельный)
19. Принцип моделирования крупномасштабного производства в уменьшенном масштабе в лабораторных условиях, используемый для проведения валидационных исследований по инактивации и (или) элиминации вирусов применим и в отношении трансмиссивных агентов губчатой энцефалопатии.
20. Производители должны представлять данные по выходу готового продукта, качеству и составу препарата крови или промежуточных продуктов, полученные в уменьшенном масштабе, сопоставимые с реальным производственным процессом.
3.3. Выбор добавляемого трансмиссивного агента губчатой энцефалопатии
21. При попадании трансмиссивного агента губчатой энцефалопатии в организм животных в экспериментальных условиях наибольшее количество инфицирующих частиц обнаруживается в крови с присутствием возбудителя у половины животных в плазме, а у другой половины животных - в лейкоцитарной пленке. Содержание инфекционных частиц в плазме в 10 000 раз ниже, чем обнаруживается в ткани мозга животных, что определяет ее наиболее пригодным материалом для проведения валидационных исследований. Максимальное содержание трансмиссивных агентов в добавляемом материале для экспериментальных исследований не должно превышать 10% от общего объема.
22. Основными факторами влияющими на выбор добавляемого трансмиссивного агента губчатой энцефалопатии, на которые необходимо обратить внимание, являются:
трансмиссивный агент губчатой энцефалопатии в добавляемом материале и вид животного, от которого он был получен;
физико-химические свойства трансмиссивного агента губчатой энцефалопатии в добавляемом материале.
Вид животного и трансмиссивный агент губчатой энцефалопатии в добавляемом материале
23. Выбор трансмиссивного агента губчатой энцефалопатии в добавляемом материале зависит от следующих факторов:
источника получения;
наличия метода количественной оценки;
схожести инфицирующих свойств с потенциальным возбудителем, присутствующим в плазме крови человека.
Получение материала от больных с вариантной болезнью Крейтцфельда - Якоба ограничено по этическим и иным причинам и его использование необязательно. Использование тканей мозга крупного рогатого скота ограничено в связи со сложностью получения материала надлежащего качества и сложностью оценки количественного содержания трансмиссивных агентов губчатой энцефалопатии.
24. Для подтверждения удаления в ходе производственного процесса не только добавляемых трансмиссивных агентов, но и возбудителя вариантной болезни Крейтцфельда - Якоба, целесообразно использовать лабораторные штаммы трансмиссивных агентов губчатой энцефалопатии (например, скрепи, наследственной болезни Крейтцфельда - Якоба, губчатой энцефалопатии крупного рогатого скота или вариантной болезни Крейтцфельда - Якоба).
25. Поскольку патогенные свойства и характеристики штаммов отличаются, для исследований необходимо использовать несколько штаммов. Методы индикации указанных лабораторных штаммов доступны. Необходимо представлять обоснование выбора используемых лабораторных штаммов.
Физико-химические свойства трансмиссивного агента губчатой энцефалопатии в добавляемом материале
26. Физико-химические свойства трансмиссивных агентов губчатой энцефалопатии до конца не изучены, несмотря на то, что их инфицирующая способность доказана в экспериментальных исследованиях на животных. Ткань головного мозга животных признана приемлемым источником накопления трансмиссивных агентов губчатой энцефалопатии.
27. В настоящее время в качестве основных рассматриваются четыре типа материалов, получаемых из гомогената головного мозга хомячка:
а) неочищенный гомогенат головного мозга. Согласно опубликованным научным данным, неочищенные гомогенаты ткани головного мозга содержат наиболее высокую концентрацию трансмиссивных агентов. Гомогеность и доступность получения указанного материала способствует выбору его для валидационных исследований. Присутствие в материале трансмиссивных агентов разных размеров позволяет использовать физические методы их удаления;
б) микросомальная фракция ткани головного мозга. Микросомальные фракции получают путем центрифугирования гомогенатов ткани головного мозга с отделением и удалением агентов крупных размеров. Остающиеся микросомальные фракции содержат трансмиссивные агенты на клеточных мембранах. Уровень их способности вызывать заболевание ниже, чем у неочищенных гомогенатов головного мозга, но достаточен для включения в валидационные исследования;
в) кавеолоподобные домены мембраны (CLD). Материал получают ультрацентрифугированием лизированных гомогенатов тканей головного мозга;
г) очищенный белок PrPSc. Очищенный белок PrPSc получают последовательной экстракцией гомогенатов головного мозга с последующим солевым осаждением и ультрацентрифугированием. Неочищенный гомогенат, микросомальная фракция и кавеолоподобные домены мембраны (CLD) ткани головного мозга могут быть использованы для валидационных исследований стадии осаждения.
3.4. Выбор методов количественного анализа
28. Оценка способности трансмиссивных агентов вызывать развитие заболевания губчатой энцефалопатией в настоящее время является «золотым стандартом» подобных исследований. Присутствие трансмиссивного агента в ткани или жидкости подтверждается развитием неврологического заболевания у лабораторного животного по окончании инкубационного периода, а также методом титрования до конечной точки.
29. Длительность инкубационного периода также используется для оценки инфицирующей способности исследуемого материала совместно с определением инфекционности методом титрования до конечной точки. Существование видовых и штаммовых различий передачи заболевания ограничивает использование материалов для инфицирования.
Например, материалы, содержащие трансмиссивные агенты спорадической болезни Крейтцфельда - Якоба человека редко используют для инфицирования диких мышей, при этом они подходят для трансгенных мышей. Указанный факт необходимо учитывать при выборе добавляемого трансмиссивного агента.
30. Биологические методы анализа длительны при воспроизведении, что связано с длительностью инкубационного периода и возможностью получения результатов по окончании 6-9 месяцев наблюдений и клинического мониторинга инфицированных животных (например, хомячков линии 263К) и до 15 - 18 месяцев нетрансгенных мышей.
31. Биологические методы должны воспроизводиться в специально оборудованных помещениях для животных с соблюдением правил работ с микроорганизмами соответствующей группы патогенности.
32. В настоящее время стандартный тест in vitro для определения присутствия трансмиссивных агентов и оценки их количественного содержания отсутствует. Несколько клеточных линий (N2a, GT1) являются неустойчивыми и могут быть инфицированы отдельными лабораторными штаммами губчатой энцефалопатии, адаптированными к мышам, при этом некоторые штаммы, трансфицированные геном РгР, могут дублировать отдельные штаммы скрепи.
33. Метод обнаружения белка PrPSc является альтернативным методом количественного определения трансмиссивных агентов губчатой энцефалопатии. Экспериментально установлено, что трансмиссивные агенты губчатой энцефалопатии состоят из белка с конформационной матрицей PrPSc, физико-химические свойства которого до сих пор не установлены. Белок, имеющий патогенную конформацию (PrPSc), относительно устойчив к протеиназе К (может конформироваться в протеаза-устойчивый белок PrPres) и денатурирующим агентам в разных концентрациях, таким как гуанидина гидрохлорид.
3.5. Выбор стадий производства
34. Учитывая устойчивость трансмиссивных агентов губчатой энцефалопатии к традиционным методам инактивации вирусов (например, термическая обработка), для исследования необходимо выбирать такие стадии производства, в ходе которых можно ожидать частичное удаление или разделение трансмиссивных агентов. Для проведения валидационных исследований стадии обработки растворителем-детергентом и термической обработки не выбираются.
35. Такие этапы производственного процесса, как фракционирование этанолом, осаждение, хроматография и фильтрация по данным разных исследователей показали значительную эффективность в удалении трансмиссивных агентов губчатой энцефалопатии.
36. Производители должны критически оценивать производственные процессы в соответствии с положениями настоящей главы.
3.6. Интерпретация данных и ограничения по проведению исследований
37. Валидационные исследования по оценке способности процесса производства удалять трансмиссивные агенты губчатой энцефалопатии имеют следующие ограничения:
а) процесс моделирования полномасштабного производства может быть несовершенным. Технологические этапы, основанные на физическом разделении, трудно смоделировать в лабораторном масштабе при проведении валидационных исследований. Особенно это касается этапа фракционирования этанолом, который вносит значительный вклад в удаление трансмиссивных агентов губчатой энцефалопатии;
б) общий вклад в удаление трансмиссивных агентов губчатой энцефалопатии необходимо оценивать суммарно для не менее двух эффективных стадий процесса производства, но такой подход не применим в случае использования разных добавляемых агентов;
в) предварительная обработка может влиять на степень очистки от добавляемого агента. Например, если исследуемый материал обработан детергентом, он может пройти следующий этап, такой как фильтрация, гораздо легче, чем необработанный;
г) методы количественного определения добавляемых агентов длительные, трудоемкие и несовершенные;
д) трансмиссивный агент губчатой энцефалопатии и вид животного, от которого он был получен, определяют выбор метода количественного определения. Несмотря на отсутствие доказательств существенного влияния происхождения добавляемого материала с трансмиссивным агентом губчатой энцефалопатии на этапы удаления, существует вероятность того, что степень его удаления зависит от происхождения добавляемого материала;
е) физико-химические свойства добавляемого трансмиссивного агента губчатой энцефалопатии могут влиять на процесс удаления. В настоящее время физико-химические свойства трансмиссивных агентов губчатой энцефалопатии не определены. Имеются доказательства, что различные добавляемые материалы с мембраносвязанным трансмиссивным агентом губчатой энцефалопатии удалялись одинаково на всех изученных этапах осаждения. В отличие от этого, добавляемые материалы с немембраносвязанным трансмиссивным агентом губчатой энцефалопатии удавалось удалять только на определенных этапах осаждения;
ж) содержание трансмиссивных агентов в крови экспериментального животного может быть низким, а в добавляемом материале максимально высоким. Существует предположение, что удаление добавляемого материала проходит менее эффективно при низких концентрациях трансмиссивного агента, чем при высоких;
з) оценка процессов инактивации и (или) удаления вирусов включает в себя оценку робастности процесса (например, изучение влияния изменений параметров производственного процесса). Сложность проведения подобных исследований с трансмиссивными агентами губчатой энцефалопатии связана с отсутствием возможности проведения повторных исследований.
38. В связи с этим, достоверность оценки удаления трансмиссивного агента губчатой энцефалопатии на каком-либо этапе производственного процесса ниже, чем при изучении возможности удаления модельного вируса.
3.7. Повторная оценка степени очистки от трансмиссивных агентов губчатой энцефалопатии
39. В случае внесения существенных изменений в производственный процесс возможно потребуется проведение повторных валидационных исследований по оценке степени очистки от трансмиссивных агентов губчатой энцефалопатии. При этом допускается использование новых научных разработок в области исследования методов количественной оценки трансмиссивных агентов in vitro, отличных от методов приведенных в настоящей главе.
3.8. Санитарная обработка оборудования
40. Образцы, содержащие возбудитель губчатой энцефалопатии длительное время сохраняют патогенность в окружающей среде в связи с трудно поддающейся инактивацией инфекционности возбудителя. Использование большинства традиционных методов дезактивации для инактивации прионов (например, использование алкилирующих агентов и детергентов) недостаточно эффективно.
41. Только отдельные традиционные методы дезактивации являются достаточно эффективными (например, вымачивание в растворе отбеливателя с концентрацией > 2% или 1-2 Н растворе натрия гидроксида (NaOH) в течение 60 мин., автоклавирование при температуре 134 - 138°С при определенном режиме поддержания давления и времени экспозиции).
42. Отдельные процедуры, признанные эталонными методами или рекомендованные ВОЗ применяются для обработки медицинских изделий или отходов производства. Применение таких процедур для производства биологических лекарственных препаратов имеет ряд ограничений. Большинство методов являются довольно жесткими по своему воздействию и могут разрушить большинство биологических лекарственных препаратов, вызвать коррозию производственного оборудования или иметь недостаточную эффективность в отношении других инфекционных агентов (например, раствор NaOH считается неэффективным в отношении спор). Использование некоторых методов обработки (например, щелочные очистители, протеазы и т.д.) находится на этапе экспериментальной оценки пригодности их использования для очистки и дезактивации.
43. В связи с устойчивостью возбудителей губчатой энцефалопатии к инактивации и способностью к прикреплению к нержавеющей стали и другим материалам, необходимо оценивать вклад процедур санитарной обработки и очистки в процессы инактивации или удаления возбудителей губчатой энцефалопатии. Необходимо оценивать влияние процедур санитарной обработки и регенерации хроматографических колонок на снижение инфекционности возбудителей губчатой энцефалопатии. Большинство процессов фракционирования заканчиваются глубинной фильтрацией и удалением использованного фильтра. В случае если указанная стадия признана эффективной, риск контаминации лекарственного препарата любыми другими инфекционными источниками, связанными с оборудованием также снижается.
44. Создание модели для изучения инактивации или удаления трансмиссивных агентов губчатой энцефалопатии, прикрепленных к металлическим поверхностям затруднено. В стандартной модели стальную проволоку погружают в образец, содержащий трансмиссивный агент губчатой энцефалопатии, и имплантируют в мозг чувствительного животного, у которого впоследствии развивается заболевание.
45. Изучение влияния санитарной обработки на снижение инфекционности на указанной модели возможно при погружении стальной проволоки в образцы с разными разведениями трансмиссивного агента до и после проведения обработки оборудования.
46. Несмотря на то, что экспериментальные растворы для дезинфекции (такие как раствор NaOH) используются для иных вариантов очистки производственного оборудования, подход с прямым переносом данных опубликованных при изучении новых методов дезинфекции медицинского оборудования, контаминированного прионами не применим без дополнительных исследований в отношении оборудования промышленного фармацевтического производства.
47. Установлено, что обработка 0,1 М раствором NaOH превращает белок PrPSc в протеаза-чувствительную форму как в растворе так и на металлической поверхности. Представленные результаты необходимо подтверждать в исследованиях по оценке инфекционности возбудителя.
48. Исходя из положений пунктов 40 - 47 настоящей главы в настоящее время не существует единых надежных указаний по санитарной обработке промышленного оборудования, используемого для переработки плазмы.
49. Все производители препаратов крови должны критически анализировать производственные процессы в соответствии с положениями настоящей главы. Выбор мер по удалению трансмиссивных агентов губчатой энцефалопатии необходимо осуществлять непосредственно для конкретного производственного процесса и моделирования наихудших возможных условий.
Глава 25. Указания по оценке иммуногенности терапевтических белков
1. Общие положения
1. Количество белков, используемых в качестве терапевтических лекарственных препаратов, неуклонно растет.
Для целей настоящей главы используется понятие, которое означает следующее:
«терапевтический белок» - белки, полипептиды и их производные, полученные с использованием рекомбинантных или нерекомбинантных систем экспрессии.
2. В целом, большинство нежелательных реакций (побочных эффектов) при применении терапевтических белков связано с фармакологическими эффектами терапевтических белков. Одним из исключений является способность терапевтических белков индуцировать нежелательный иммунный ответ. Риск иммуногенности варьирует между отдельными препаратами и группами лекарственных препаратов, с одной стороны, и между отдельными пациентами и группами пациентов, с другой стороны. В настоящей главе приводится перечень вопросов по иммуногенности, подлежащих рассмотрению в резюме модуля 2 регистрационного досье лекарственного препарата. Данное резюме отражает обоснованность подхода по оценке риска относительно иммуногенности, подтверждает, что объем и тип исследований иммуногенности до регистрации лекарственного препарата и программа пострегистрационных исследований, включенных в план управления рисками, составлены с учетом риска иммуногенности и серьезности ее потенциальных или наблюдаемых клинических последствий.
3. С регуляторной точки зрения прогностическая значимость результатов исследований на животных для оценки иммуногенности биологического лекарственного препарата для человека является низкой в связи с различием между иммунной системой человека и животных и неизбежным возникновением у животных иммунного ответа к белкам человека. Разработка адекватных скрининговых и подтверждающих методов для изучения иммунного ответа на терапевтический белок является ключевым моментом в оценке иммуногенности. Заявители должны продемонстрировать, что методы определения антител применимы для доказательства корреляций выявленных индуцированных антител с клиническими последствиями.
4. Целью исследований иммуногенности является выявление иммунного ответа на терапевтический белок и его влияния на клинические последствия. Таким образом, оценка иммуногенности должна основываться на комплексном анализе иммунологических, фармакокинетических, фармакодинамических показателей, а также данных клинической эффективности и безопасности лекарственного препарата. Вопросы иммуногенности должны быть отражены в плане управления рисками.
5. Положения настоящей главы, включающие в себя общие подходы к изучению иммуногенности, необходимо адаптировать к программе фармацевтической разработки конкретного вида биологического лекарственного препарата. В соответствии с пунктом 26 Правил регистрации и экспертизы заявитель вправе обратиться за научной консультацией в уполномоченные органы (экспертные организации) государств-членов для такой адаптации.
6. Терапевтические белки распознаются иммунной системой человека. Вслед за распознаванием часто на них формируется иммунный ответ. Этот потенциально опасный иммунный ответ является комплексным и, помимо образования антител к лекарственному препарату, включает активацию Т-клеток и ответ врожденной системы иммунитета.
7. Последствия иммунного ответа на терапевтический белок варьируют от кратковременного транзиторного появления антител без каких-либо клинически значимых явлений до тяжелых, угрожающих жизни состояний. Потенциальные клинически значимые последствия развития нежелательного иммунного ответа включают снижение эффективности терапевтического белка, тяжелые острые иммунные реакции, такие как анафилаксия, и для терапевтических белков, применяемых в качестве заместительной терапии - перекрестную реактивность с эндогенным белком аналогом.
8. На иммуногенность терапевтических белков оказывают влияние множественные факторы, которые делятся на факторы зависящие от пациента и факторы опосредованные заболеванием или лекарственным препаратом. Зависящие от пациента факторы могут предрасполагать к развитию иммунного ответа у субъекта, к ним относятся: генетические особенности (наследственная предрасположенность), пред существующий иммунитет, иммунный статус, включая терапию иммуномодулирующими лекарственными препаратами. К факторам, связанным с лечением, относятся режим дозирования и путь введения лекарственного препарата. Факторы, опосредованные лекарственным препаратом, которые влияют на вероятность развития иммунного ответа, включают в себя характеристики лекарственного препарата, обусловленные производственным процессом, составом препарата и его стабильностью.
9. В зависимости от иммуногенного потенциала лекарственных препаратов, содержащих терапевтический белок и (или) частоты распространенности заболевания, объем данных по изучению иммуногенности до регистрации может быть ограничен. Контролируемые клинические испытания не позволяют полностью оценить редкие нежелательные реакции, эффекты или медленно развивающиеся иммунные реакции. После регистрации требуется дальнейшая систематическая оценка иммуногенности, которую необходимо предусмотреть в плане управления рисками.
2. Сфера применения
10. Общие указания, изложенные в настоящей главе, преимущественно касаются вопросов, связанных с установлением факта развития нежелательного иммунного ответа на очищенный терапевтический белок у пациентов, и способов его систематической оценки. Положения настоящей главы распространяются на терапевтические белки, а также лекарственные препараты, в которых терапевтические белки являются компонентами (например, конъюгатами).
11. Положения настоящей главы не распространяются на препараты факторов свертывания крови, вакцины или лекарственные препараты гетерогенных иммуноглобулинов и человеческих иммуноглобулинов, выделенных из плазмы крови.
3. Факторы, влияющие на развитие иммунного ответа на терапевтический белок
3.1. Факторы, зависящие от пациента или опосредованные заболеванием
Генетические факторы, модулирующие иммунный ответ
12. Генетические факторы могут влиять на иммунные реакции, развивающиеся в ответ на терапевтический белок, и могут быть причиной межиндивидуальной вариабельности ответа. Генетические различия на уровне молекул главного комплекса гистосовместимости (ГКГ) и Т-клеточного рецептора могут модифицировать процесс распознавания антигена, тогда как генетические особенности на уровне модулирующих факторов, таких как цитокины и рецепторы цитокинов, могут влиять на длительность и интенсивность иммунного ответа.
Генетические факторы, обусловленные дефектом генов
13. В случае, когда терапевтический белок применяется в качестве препарата замещения эндогенного белка для пациента, у которого выявлена полная или частичная недостаточность эндогенного белка, или он является носителем модифицированной формы природного аналога, физиологический (неизмененный) антиген препарата может восприниматься как нео-антиген и иммунная система пациента будет распознавать терапевтический белок как чужеродный.
Факторы, связанные с возрастом
14. Данные по оценке иммуногенности, полученные в одной возрастной группе, не всегда экстраполируются на пациентов других возрастных групп, поскольку иммунный ответ на терапевтические белки может зависеть от возраста пациента. В педиатрической популяции отмечается различный уровень созревания иммунной системы в зависимости от возраста, и ожидается развитие различных иммунных реакций на биологический лекарственный препарат.
15. Если лекарственный препарат предназначен для применения в педиатрической популяции, клинические исследования проводят с участием пациентов соответствующей возрастной группы или групп. Если лекарственный препарат предназначен для применения у пожилых людей, необходимо учитывать вероятность изменения иммунного ответа у таких пациентов.
Факторы, опосредованные заболеванием
16. Заболевание пациента само по себе может быть важным фактором развития нежелательного иммунного ответа. Пациенты с активированной иммунной системой (например, страдающие хроническими инфекциями, аллергией и аутоиммунными воспалительными заболеваниями) могут быть более склонны к развитию иммунного ответа на терапевтические белки.
17. При других состояниях (например, при истощении вследствие дефицита питания, прогрессировании онкологического заболевания, поздних стадиях и распространенности ВИЧ-инфекции, органной недостаточности) развитие иммунного ответа на введение терапевтического белка менее вероятно вследствие нарушения функции иммунной системы.
18. В отношении некоторых лекарственных препаратов известно, что возможность развития гуморального иммунного ответа может различаться в зависимости от терапевтических показаний к применению или стадии заболевания. Гуморальный ответ на лекарственный препарат также может быть изменен на фоне развития вирусной инфекции у пациента.
Сопутствующая терапия
19. Сопутствующая терапия может либо снижать, либо повышать риск развития иммунного ответа на терапевтический белок. Как правило, иммунная реакция на терапевтический белок снижается при одновременном применении иммунодепрессантов. Иммунный ответ на терапевтический препарат является результатом взаимодействия многих факторов (например, предшествующего или сопутствующего облучения (радиационной терапии) или уровня экспозиции препарата), поэтому выводы о потенциальном влиянии одновременного применения иммуномодулирующих лекарственных препаратов не являются однозначными. Необходимо учитывать предыдущее лечение, которое может влиять на иммунные реакции и иммунную систему. Если клинические исследования лекарственного препарата с новой активной фармацевтической субстанцией были выполнены в комбинации с иммунодепрессантами, при утверждении показаний к применению белкового препарата в виде монотерапии должны быть представлены адекватные клинические данные о профиле иммуногенности препарата при его назначении в отсутствии терапии иммунодепрессантами.
Факторы, связанные с терапией
20. На развитие иммунного ответа на терапевтический белок могут влиять режим дозирования, доза и путь введения лекарственного препарата. Лекарственные препараты, вводимые внутривенно, могут быть менее иммуногенными, чем лекарственные препараты, вводимые подкожно или внутримышечно. Ингаляционное, внутрикожное, а также внутриглазное введение может также усиливать иммунные реакции, развивающиеся в ответ на терапевтический белок.
21. При краткосрочном лечении, вероятность развития нежелательного иммунного ответа ниже, чем при долгосрочном лечении. Повторное назначение лекарственного препарата после длительного перерыва может быть связано с усиленным иммунным ответом.
Предсуществующие антитела
22. Пред существующие антитела представляют собой эндогенные антитела, которые являются специфичными перекрестно-реагирующими, направленными к эпитопам белков или гликанов, способными взаимодействовать с эпитопами терапевтических белков. Предсуществующие антитела могут формироваться в результате лечения лекарственными препаратами на основе аналогичных или родственных белков, но также могут выявляться у ранее не получавших лечение пациентов. Точное происхождение таких антител в большинстве случаев неизвестно.
3.2. Факторы, связанные с лекарственным препаратом
23. Важные факторы, влияющие на иммуногенность терапевтических белков, включают в себя:
происхождение (например, чужеродное или человеческое) и природу активной фармацевтической субстанции (эндогенные белки, посттрансляционные модификации);
значительные модификации терапевтического белка (например, пегилирование и белки слияния (fusion proteins));
родственные примеси, связанные с продуктом (например, продукты деградации, родственные соединения, агрегаты);
примеси, связанные с процессом производства (белки, липиды или ДНК клеток-хозяина, микробные контаминанты);
состав (вспомогательные вещества) и взаимодействие лекарственного препарата и (или) вспомогательных веществ с материалом первичной упаковки (например, контейнеры, пробки).
Структура белка и посттрансляционные модификации
24. Иммунологическая толерантность к эндогенным белкам вариабельна (как правило, толерантность слабее к белкам с низким уровнем содержания (низкодозовая толерантность), чем к белкам с большим содержанием). Например, уровни цитокинов и факторов роста являются низкими, поэтому выявление аутоантител к цитокинам и факторам роста у здоровых людей не является редкостью.
25. Терапевтические белки являются аналогами эндогенных белков человека и могут вызывать развитие иммунного ответа из-за изменений в аминокислотной последовательности или изменений в структуре белка по сравнению с эндогенным белком, что является результатом посттрансляционных модификаций или других изменений на любом этапе процесса производства активной фармацевтической субстанции и (или) лекарственного препарата, при его хранении или применении.
26. Т-клеточные эпитопы представляют собой короткие линейные пептиды, в которых за счет модификации могут возникать различия в аминокислотной последовательности между эндогенным и терапевтическим белком. Соответственно, проведение исследований по идентификации потенциальных Т-клеточных эпитопов необходимо для выбора новых белков или пептидов в целях разработки на их основе лекарственного препарата.
27. Особенности гликозилирования могут влиять как на физикохимические, так и на биологические свойства белка. Присутствие или отсутствие олигосахаридных групп, а также структура углеводных фрагментов могут оказывать как прямое, так и косвенное влияние на иммуногенность терапевтических белков. Гликаны сами по себе могут индуцировать иммунный ответ (например, гликаны нечеловеческого происхождения), или их присутствие может влиять на конформацию белка таким образом, что белок становится иммуногенным.
28. Химически модифицированные белки представляют собой новые активные фармацевтические субстанции, способные инициировать иммунный ответ. Были выявлены индуцированные специфические антитела, направленные против полиэтиленгликолевой части пегилированных (ПЭГ) белков, включая предсуществующие антитела против полиэтиленгликолевой части пегилированных белков. Пэгилирование и гликозилирование с другой стороны также способны снижать иммуногенность терапевтического белка, путем экранирования иммуногенных эпитопов, сохраняя при этом нативную конформацию белка.
29. Не-аналоговые терапевтические белки, такие как слитые белки (белки слияния (fusion proteins)), могут содержать нео-эпитопы из-за введения чужеродных пептидных последовательностей, (например, в линкерах (участках соединения)). Белки слияния, состоящие из чужеродного и собственного белка, также как химерные белки, требуют особого внимания и настороженности из-за потенциального присутствия чужеродного фрагмента, способного провоцировать формирование иммунного ответа на собственный белок (распространение эпитопа (epitope-spreading)). В таких случаях проводится идентификация антигенного компонента белка слияния, что важно в плане оценки риска иммуногенности.
Состав и упаковка
30. Состав вспомогательных веществ, помимо безопасности для пациента, подбирается с целью наилучшего поддержания нативной конформации терапевтического белка. Выбор оптимального и стабильного состава зависит от понимания физической и химической природы активной фармацевтической субстанции (действующего вещества), самих вспомогательных веществ и их взаимодействия в комбинации друг с другом и с материалом первичной упаковки (например, выщелачивание и примеси из контейнеров и укупорочных материалов, зависящие от производственного процесса их получения, вольфрам). Состав и происхождение вспомогательных веществ и материалов первичной упаковки могут влиять на иммуногенность терапевтических белков. Этот фактор необходимо учитывать при внесении изменений в процесс производства лекарственного препарата на этапе его первичной упаковки.
31. Условия клинического применения лекарственного препарата (например, разведение в инфузионных растворах и использование инфузионного оборудования, произведенного из различных материалов) могут влиять на качество препарата и оказывать негативное воздействие, способствующее проявлению иммуногенности препарата.
Агрегация и образование аддуктов
32. Денатурация и агрегация терапевтического белка потенциально могут вызвать иммунный ответ. Агрегация и образование аддуктов белков могут приводить к оголению (появлению) новых эпитопов или образованию поливалентных эпитопов, которые способны стимулировать иммунную систему. Кроме того, агрегация может усилить специфический иммунный ответ на белок и вызвать образование индуцированных антител. Процесс очистки, состав и условия хранения, среди прочих факторов, могут приводить к образованию агрегатов или аддуктов. Результаты доклинических исследований in vivo свидетельствуют о том, что удаление агрегатов (присутствующих в виде видимых или невидимых частиц) способствует снижению иммуногенности.
33. Агрегаты с более высокой молекулярной массой более склонны к индукции иммунного ответа, чем агрегаты с более низкой молекулярной массой. Кроме того, повторяющиеся упорядоченные эпитопы (поливалентные эпитопы), которые часто формируются при образовании белковых агрегатов (например, вирус-подобные частицы), могут непосредственно активировать В-клетки. Обширное перекрестное сшивание рецепторов В-клеток структурами более высокого порядка может активировать В-клетки и стимулировать продукцию антител не только к агрегированной, но и к мономерной форме белка.
Примеси
34. Выделяют ряд потенциальных примесей, присутствующих в активной фармацевтической субстанции и лекарственном препарате терапевтических белков, которые потенциально могут выполнять роль адъювантов (веществ, которые усиливают развитие иммунного ответа на антигены) или индуцировать иммунный ответ на себя. В качестве таких адъювантов в активной субстанции рассматриваются производственные примеси (например, белки клеток-хозяина, липиды или ДНК клеток-хозяина, микробные белки и другие контаминирующие агенты производственного процесса). Риск иммуногенности на белки клетки-хозяина зависит от источника (клеточной линии) получения терапевтического белка.
4. Возможные клинические последствия иммуногенности
35. Цель исследования иммуногенности терапевтических белков заключается в установлении ее клинической значимости (определении влияния нежелательного иммунного ответа на фармакокинетику, фармакодинамику, безопасность и эффективность лекарственного препарата). Факторы, которые определяют способность антител к терапевтическим белкам вызывать клинически значимые эффекты (последствия), включают в себя распознаваемый эпитоп, аффинность, класс иммуноглобулина антител. Влияние на клинический результат оказывает способность иммунных комплексов активировать комплемент.
4.1. Влияние на эффективность
36. Антитела могут оказывать влияние на эффективность терапевтического белка либо путем воздействия на фармакодинамическое взаимодействие между терапевтическим белком и его мишенью, либо путем изменения его фармакокинетического профиля.
37. Когда антитела связываются с активным сайтом (или расположенными вблизи него сайтами) антигена терапевтического белка или вызывают конформационные изменения, связывание терапевтического белка с соответствующими рецепторами может быть подавлено. Такие антитела определяют как нейтрализующие антитела.
38. Антитела могут изменять экспозицию (воздействие) терапевтического белка путем увеличения или уменьшения клиренса терапевтического белка. Когда воздействие лекарственного препарата снижается из-за увеличения клиренса или увеличивается, эти антитела обозначают как элиминирующие (очищающие) и сохраняющие антитела, соответственно. Антитела, индуцированные введением терапевтического белка, могут обладать как нейтрализующими, так и элиминирующими (очищающими) или сохраняющими (задерживающими выведение из организма лекарственного препарата) свойствами.
39. Предполагается, что антитела, не влияющие на клиренс (не-элиминирующие) и не-нейтрализующие, будут оказывать меньшее влияние на клинические последствия, связанные с эффективностью лекарственного препарата. Эффекты индуцированных антител на клинические последствия терапевтических белков варьируются от отсутствия влияния до полной потери эффективности.
40. Предварительное применение подобных или родственных белковых препаратов, приводящее к формированию иммунных реакций (предсуществующая реактивность), может модифицировать ответ на новый терапевтический белок (повлиять на фармакокинетику, безопасность или эффективность).
41. Последствия формирования таких антител могут быть негативными. Для пациентов, получающих биологические лекарственные препараты в качестве заместительной терапии (например, при введении препаратов факторов свертывания крови или препаратов фермент-заместительной терапии предсуществующие антитела могут перекрестно реагировать с белками вновь введенного биологического лекарственного препарата, устраняя его эффект). Поэтому необходимо учитывать потенциальную перекрестную реактивность терапевтического белка с предсуществующими антителами.
4.2. Влияние терапевтических белков на безопасность лекарственного препарата
42. В целом, наиболее неблагоприятные эффекты терапевтических белков связаны с их фармакологическими эффектами. Основное исключение состоит в том, что иммунные реакции могут приводить к неблагоприятным последствиям. Иммуно-опосредованные нежелательные реакции могут быть как острыми, так и замедленными.
43. Менее серьезные иммуно-опосредованные нежелательные реакции включают в себя инъекционные и инфузионные реакции. Неаллергические (не связанные с формированием IgE-антител) инфузионные реакции наблюдаются во время первых инфузий и могут быть смягчены предварительным назначением соответствующих лекарственных препаратов (проведение премедикации).
Гиперактивные (острые) реакции
44. Гиперактивные (острые) реакции, связанные с инфузией, (острые инфузионные реакции, включая анафилактические (анафилактоидные реакции (тип I)), могут развиваться в течение нескольких секунд или через несколько часов после инфузии лекарственного препарата.
45. Все острые инфузионные реакции потенциально связаны с формированием иммунного ответа. Часть из них являются аллергическими (анафилактическими) реакциями по своей природе и как правило определяются выработкой иммуноглобулина Е (IgE), но ряд инфузионных реакций не являясь истинными аллергическими реакциями (анафилактоидные реакции) по своим клиническим проявлениям могут быть подобными анафилактическим реакциям. Гиперактивные (острые) реакции могут сопровождаться выраженной гипотензией, бронхоспазмом, отеком гортани или глотки, затруднением дыхания и (или) крапивницей. Предсуществующий иммунитет может изменить безопасность терапевтического белка (например, приводить к увеличению частоты и (или) тяжести реакций гиперчувствительности).
Отсроченные реакции (реакции замедленного типа)
46. В дополнение к гиперактивным (острым) реакциям необходимо учитывать возможность развития гиперчувствительности замедленного типа (опосредованной Т-клетками) и реакций, опосредованных иммунными комплексами. Риск развития таких реакций возрастает с увеличением интервала между введением лекарственного препарата или при многократной замене лекарственных препаратов, относящихся к одной группе. Такие реакции замедленной гиперчувствительности необходимо четко отличать от инфузионных реакций. Заявители должны обеспечить систематический сбор данных об отсроченных клинических последствиях применения терапевтического белка. К клиническим проявлениям таких реакций относят миалгию, артралгию с повышением температуры тела, кожную сыпь, зуд.
Аутоиммунные реакции при перекрестной реактивности биологического препарата с эндогенными аналогами
47. Опасным для жизни клиническим последствием образования антител против терапевтического белка является их перекрестная реактивность с эндогенным белком, когда этот белок играет ключевую роль в физиологических функциях и не имеет избыточной продукции для выполнения этой роли. Например, антитела, перекрестно реагирующие с эндогенным эритропоэтином, были причиной развития истинной красно-клеточной аплазии у пациентов с почечной недостаточностью, получавших препарат эпоэтина альфа. Антитела, индуцированные введением лекарственных препаратов, созданных на основе новых конструкций (например, слитых белков (fusion proteins), содержащих физиологически функциональные молекулы), необходимо исследовать на перекрестную реактивность с соответствующими эндогенными белками.
5. Доклиническая оценка иммуногенности и ее последствий
48. Терапевтические белки в большинстве случаев проявляют видовую специфичность (белки человека распознаются организмом животного как чужеродные белки). В связи с этим прогностическая значимость доклинических исследований на животных по оценке иммуногенности терапевтического белка считается низкой. Проведение доклинических исследований in vitro или in vivo, направленных на прогнозирование иммуногенности у человека, не требуется (если не обосновано иное).
49. Необходимо уделять постоянное внимание возможности появления новых технологий (новые модели in silico, in vitro и in vivo), которые могут быть использованы в качестве инструментов во время разработки препарата или для первой оценки риска иммуногенности при клиническом исследовании. Методы in vitro, основанные на использовании клеток врожденной и адаптивной системы иммунитета, могут быть полезны для выявления клеточно-опосредованного иммунного ответа.
50. Проблемы иммуногенности могут возникать из-за присутствия в препарате примесей или контаминантов. Следует полагаться на процессы очистки для удаления примесей и контаминантов, а не на разработку программы доклинических исследований для их идентификации и характеристики. Ожидается, что при проведении клинических исследований, в которых оценивается иммуногенность, будут получены материалы, достаточно убедительные для лекарственного препарата, предназначенного для регистрации.
51. Изучение формирования антител к лекарственному препарату в исследованиях на животных может быть проведено в рамках исследований хронической токсичности. Для интерпретации результатов этих исследований необходимо использовать рекомендации, изложенные в документе ICH S6 (R1) и настоящих Правилах. Когда изучение и характеристика индуцированных антител не являются частью протокола исследования, взятые образцы крови необходимо хранить для последующей оценки, результаты которой необходимы для интерпретации результатов проведенных доклинических исследований. Используемые методики должны быть валидированы. При исследовании токсичности, где в образцах обычно присутствуют высокие концентрации терапевтического белка, необходимо учитывать интерференцию терапевтического белка на уровни определяемых антител. Оценка иммуногенности при проведении исследования токсичности одной дозы (острая токсичность) не требуется. Для однократной дозы при фармакокинетическом исследовании оценка антител актуальна.
52. Иммунный ответ на терапевтический белок, являющийся аналогом эндогенного белка, может привести к появлению перекрестно реагирующих антител, направленных против эндогенного белка, в тех случаях, когда синтез последнего сохраняется. Как правило, если риски безопасности предсказуемы на основании имеющейся информации о биологических функциях эндогенного белка, исследования на животных для подтверждения этих рисков не требуются. При отсутствии достаточной информации и при наличии имеющихся теоретических предпосылок, указывающих на возможный риск в отношении безопасности применения препарата, для получения информации о возможных последствиях нежелательного иммунного ответа необходимо проведение исследований с иммунизацией животных терапевтическим белком или гомологичным белком соответствующего вида животных. Данные, полученные на животных моделях, о последствиях индукции иммунного ответа на эндогенный белок или их отсутствии (дисфункции), необходимо отразить в сводном резюме по иммуногенности.
53. Нежелательная иммуногенность биологических лекарственных препаратов может проявляться как в виде гуморального, так и клеточного иммунного ответа. При формировании клеточного иммунного ответа фармакодинамические или побочные эффекты (или ожидаемые эффекты) опосредуются иммунными клетками. О развитии клеточного ответа могут свидетельствовать реакции гиперчувствительности замедленного типа или формирование цргготоксических Т-клеток.
54. При разработке биоподобных биологических лекарственных препаратов сравнение ответа по формированию антител на биоподобный и референтный препараты на модели животных, как части сравнительных исследований по доказательству их подобия, не требуется, что связано с низкой прогностической значимостью потенциальной иммуногенности таких препаратов для человека. Если в редких случаях возникает потребность в исследовании токсичности или когда проводятся фармакокинетические исследования, проводится оценка формирования антител с целью интерпретации результатов исследований.
6. Разработка методов обнаружения и определения иммунного ответа у человека
55. Разработка стратегии комплексного анализа, соответствующей предполагаемому плану лечения, имеет решающее значение для выяснения клинической значимости данных, полученных по оценке иммуногенности. Аналитические методы и методологию оценки иммунного ответа необходимо выбрать и (или) разработать до этапа клинической разработки лекарственного препарата. Основное внимание исследователей, как правило, направлено на выявление антител и их характеристику, поскольку это имеет большое значение для определения клинической значимости в плане безопасности и эффективности применения лекарственного препарата. Вместе с тем, клеточно-опосредованный иммунный ответ может также играть большую роль, поэтому заявитель должен рассматривать необходимость его оценки в индивидуальном порядке, где это применимо.
56. Несмотря на то, что методы уточняются в ходе разработки лекарственного препарата и аналитическая пригодность переоценивается в соответствии с использованием методики, заявитель должен представить всю необходимую информацию и полные данные по валидации методик, используемых для оценки иммуногенности, при подаче документов для регистрации лекарственного препарата.
57. Общая стратегия изучения гуморального иммунного ответа предусматривает необходимость использования чувствительных и валидированных методов оценки иммуногенности. Как правило, при проведении исследований используется поэтапный подход. Такой подход включает методы скрининга для идентификации образцов (пациентов) с наличием исследуемых антител и последующий этап подтверждения наличия антител, определения их специфичности и использование ряда функциональных методов для оценки нейтрализующей способности выявленных антител.
58. Любое отклонение от этой концепции должно быть надлежащим образом обосновано заявителем в рамках предрегистрационных (научных) консультаций с уполномоченным органом до подачи документов на регистрацию. Все ключевые методики, используемые для скрининга, подтверждения, определения нейтрализующих антител должны быть валидированы для предполагаемого дальнейшего использования. В некоторых случаях требуется тестирование образцов для оценки перекрестной реактивности с другими препаратами на основе одного и того же белка или эндогенного белка, если это имеет значение для оценки влияния на клиническую безопасность и эффективность.
59. При выборе методик необходимо учитывать быстро развивающиеся технологии совершенствования аналитических методик, используемых для оценки и характеристики антител. Кроме того, необходимо предусмотреть другие аналитические методы, не направленные на выявление антител, например, такие методы для определения уровня остаточного содержания препарата и оценки его клинической значимости, как методы определения имеющих значимость биомаркеров или фармакокинетических параметров, которые позволят оценить и охарактеризовать влияние индуцированных антител (если такие выявлены) на клинические эффекты (в соответствии с приложением к настоящей главе).
60. Если установлена индукция антител у пациентов, проводится оценка кинетики формирования антител и ее продолжительность, а также оценивается степень выраженности гуморального ответа, поскольку он может коррелировать с клиническими проявлениями последствий наличия антител. В таких случаях образцы сыворотки или плазмы необходимо охарактеризовать в отношении уровня антител (титр), нейтрализующей способности и, других характеристик, определяемых в каждом конкретном случае в соответствии со свойствами биологического препарата, особенностями терапии пациентов, целью исследования, клиническими симптомами и другими возможными факторами. Последующая характеристика антител, если требуется, должна включать, определение класса и подкласса антител (изотип), их аффинность и специфичность. Методики, используемые для оценки указанных характеристик, должны быть аттестованы и соответствовать их назначению.
6.1. Методы для скрининга
61. Использование скрининговых методов является первым шагом в оценке иммуногенности. Методы должны быть чувствительными и способными обнаруживать все клинически значимые антитела (включая подклассы IgM и IgG), индуцированные введением лекарственного препарата, у всех серопозитивных пациентов (то есть во всех серопозитивных образцах). Желателен низкий уровень ложноположительных результатов (предпочтительно 5%), при этом ложноотрицательных результатов не должно быть.
62. Скрининг проводится с использованием иммунологических методов, которые базируются на разнообразных способах и системах обнаружения, описанных в приложении в настоящей главе. Все методики скрининга направлены на выявление взаимодействия между антигенами и антителами (связывание), но основываются на разных научных (технических) принципах. Эти методики характеризуются достаточно высокой производительностью, выполнение соответствующих процедур автоматизировано, каждая методика имеет свои особенности и соответствующие ограничения, которые необходимо учитывать (в соответствии с главой 10 настоящих Правил).
63. Методы должны быть разработаны, подобраны, оптимизированы и валидированы в соответствии с их предполагаемым использованием. При выборе скрининговых методик необходимо учитывать все методологические проблемы и мешающие факторы, которые могут повлиять на результаты тестирования. Например, иммуноферментный анализ (ELISA), основанный на прямом связывании с антигеном, который непосредственно иммобилизован на поверхности пластиковых лунок, является самым простым для выполнения методом, при этом такой анализ характеризуется высокой частотой выявления ложноположительных результатов. Кроме того, для такого типа методов характерна высокая частота ложноотрицательных результатов при тестировании образцов, содержащих низкоаффинные антитела. Для исключения методологических проблем необходимо предусмотреть возможность использования других подходящих видов анализа, например, таких как модифицированный иммуноферментный анализ (brindging assays), электрохемилюминесценция или поверхностный плазмонный резонанс, с учетом их ограничений. При использовании некоторых скрининговых методик возможно маскирование эпитопов, что приводит к получению ложноотрицательных результатов. Данная проблема решается, например, путем маркировки детектирующих реагентов с использованием процедур, которые предотвращают маскирование определенного эпитопа (эпитопов).
64. Реагенты, используемые при выполнении анализа (например, блокирующие реагенты), необходимо тщательно изучить. Блокирующие реагенты, такие как BSA и молоко, содержат нечеловеческие гликаны, которые иногда могут присутствовать в составе белковых препаратов, полученных с использованием клеток животных, отличных от человека. В этой связи индуцированные антитела, направленные к этим гликанам, при выполнении методики анализа могут не обнаруживаться.
65. Образцы (обычно сыворотка или плазма) содержат вещества, которые могут мешать проведению анализа, оказывая матриксные эффекты, что приводит к получению ложноположительных или отрицательных результатов и (или) некорректному определению уровня содержания антител (например, компоненты комплемента или рецепторы комплемента, маннозосвязывающий белок, Fc рецепторы, растворимые молекулы-мишени и ревматоидный фактор). Влияние таких матриксных компонентов на результаты анализа необходимо рассматривать и оценивать при проведении валидации методики. Чтобы ослабить потенциальное влияние эффекта матрикса, необходимо выполнить корректирующие меры и обосновать выбранный подход, с учетом наличия ограничений соответствующих методов. Кроме того, остаточное содержание терапевтического белка (лекарственного препарата), присутствующего в крови пациента, может образовывать комплексы с индуцированными антителами и вследствие этого снижать поддающуюся обнаружению концентрацию антител. Эта интерференция может различным образом влиять на методику, в зависимости от ее разновидности, формата и типа, а также характеристики антител. Вопросы, связанные с указанными обстоятельствами, необходимо решать во время валидации методики. Если такое влияние присутствия остаточного содержания лекарственного препарата выявляется, его можно преодолеть (разрешить), используя различные методические приемы (например, путем диссоциации иммунных комплексов с помощью кислоты, удалением избыточного количества лекарственного препарата путем твердофазной абсорбции, использованием длительного периода инкубации и (или) использованием метода, позволяющего проводить анализ образцов при его высоких разведениях). В некоторых случаях допускается удалить из образца остаточное содержание лекарственного препарата или антиген-мишень, используя лектины или ксеногенные антитела. Такие подходы должны быть валидированы в отношении их эффективности и должно быть показано, что они не оказывают негативного влияния на конечные результаты анализа. В некоторых случаях интерференция остаточного содержания терапевтического белка не допускается путем отбора проб для оценки антител спустя достаточное количество времени после введения препарата, что позволяет ему элиминироваться из кровотока до отбора проб. Однако такой подход не должен значительно усложнять процесс обнаружения антител и влиять на схему лечения пациента. Заявитель должен продемонстрировать, что на чувствительность методики по выявлению антител в образцах не влияет определенный уровень терапевтического белка, который выше уровня содержания в тестируемых образцах на наличие антител. Из-за технических ограничений не всегда есть возможность разработать полностью нечувствительные методики к присутствию терапевтического белка. Необходимо использовать наилучший из возможных вариантов анализа, выбранный подход должен быть надлежащим образом обоснован.
6.2. Методы, подтверждающие наличия антител
66. Подтверждающие анализы предназначены для подтверждения положительных результатов и исключения всех ложноположительных образцов, отобранных в результате скрининга. При выборе метода необходимо учитывать ограничения и характеристики методов скрининга. Общим подходом для подтверждения наличия антител является добавление избыточного количества антигена в образец с последующим сравнением результатов анализа образцов, с добавлением и без добавления антигена, и результатами скринингового анализа. Данная процедура должна приводить к ингибированию первоначального связывания антител с антигеном и снижению частоты положительных сигналов от истинно положительных образцов.
67. Антитела, присутствующие в подтвержденных положительных образцах, должны быть исследованы на их количественное содержание (титр) и специфичность к терапевтическому белку. Известно, что антитела могут быть индуцированы также другими веществами, присутствующими в лекарственном препарате, такими как родственные соединения и посторонние примеси, то есть компонентами, связанными с продуктом или связанными с процессом производства (например, белки клетки-хозяина). В таких случаях методы по выявлению антител, направленных против этих примесей, необходимо разработать и валидировать для тестирования образцов пациентов, при этом уровень примесей необходимо свести к минимуму, чтобы избежать возможности развития на них иммунного ответа.
6.3. Методы оценки нейтрализующей способности антител
68. Нейтрализующую способность антител, присутствующих в положительных образцах, необходимо оценивать как часть изучения иммуногенности, поскольку это часто коррелирует со снижением клинического ответа на биологический препарат. Отклонение от этой концепции требует обоснования. В таких случаях заявитель вправе обратиться за консультацией в уполномоченный орган (экспертную организацию) государства-члена. Нейтрализующие антитела ингибируют биологическую активность терапевтического белка путем связывания с эпитопом (эпитопами) внутри или вблизи активного сайта (сайтов) молекулы или вызывают конформационные изменения. Поскольку нейтрализующие антитела могут непосредственно вызывать клинические эффекты, для их обнаружения требуются специфичные и чувствительные методы in vitro. В основном используются два типа методов по определению нейтрализующей активности антител - методы на основе использования клеток и методы без их использования (не-клеточные методики).
69. Необходимо использовать метод, который хорошо реагирует на биологический продукт и является толерантным к остаточному содержанию терапевтического белка. Биологические методы, используемые для тестирования специфической биологической активности терапевтического белка, часто адаптируются для оценки нейтрализующих антител. Однако, эти методы требуют совершенствования (доработки), для того чтобы оптимизировать условия оценки выявления нейтрализующей способности антител.
70. Понимание механизма действия, мишени и эффекторного пути терапевтического воздействия лекарственного препарата имеет решающее значение при выборе принципа метода для анализа нейтрализующих антител. Необходимо также учитывать клинические последствия, связанные с влиянием риска формирования нейтрализующих антител. Для лекарственных препаратов-агонистов часто используются методы, основанные на использовании клеточных культур, в то время как для лекарственных препаратов на основе гуморальных мишеней молекул-антагонистов чаще предполагается использовать методы конкурентного связывания с лигандами (CLB) без использования клеток. Для лекарственных препаратов, которые проявляют свою активность только посредством прямого связывания с другими молекулами (например, некоторые лекарственные препараты моноклональных антител (МкАТ), более подходящим является метод CLB или другие альтернативные методы. Однако при выборе для использования этих методов следует доказать, что они объективно отражают потенциальную нейтрализующую способность антител. Для лекарственных препаратов моноклональных антител являющихся антагонистами мишеней, клиническая эффективность которых обусловлена эффекторными функциями антител, используются методы на основе клеток, поскольку механизм действия препарата не может быть адекватно отражен при анализе бесклеточным методом CLB (выявление сцепленных с полом рецессивных летальных мутаций у Drosophila melanogaster).
71. При оценке нейтрализующей способности антител, как правило, выбирают одну концентрацию биологического лекарственного препарата и проводят разведения каждого исследуемого образца, определяя ингибирующее действие разведений образца, со снижением концентрации антител, на анализируемый ответ. Это позволяет определить эффект нейтрализующей дозы и рассчитать нейтрализующую способность («титр») для каждого исследуемого образца.
72. При скрининге, во время валидации метода необходимо подтвердить, что нейтрализация действительно связана с действием антител и не связана с другими ингибирующими компонентами, потенциально присутствующими в матриксе образца. С этой целью для оценки проявления специфичности антител следует рассмотреть такие подходы, как истощение антител или использование альтернативных стимулов (если результаты анализа зависят от воздействия множественных стимулов). Нейтрализующая активность не обязательно коррелирует со связыванием антител, то есть образцы, содержащие значительное или высокое количество связывающих антител, могут не нейтрализовать биологическую активность, тогда как образцы, содержащие более низкие количества связывающих антител, могут нейтрализовать определенное (в зависимости от образца) количество препарата. Нейтрализующая активность может зависеть от конкретного препарата и определяется путем проведения специальных исследований.
6.4. Стратегия оценки иммуногенности (дизайн исследований и интерпретация результатов)
73. План исследования иммуногенности должен быть тщательно разработан, чтобы обеспечить выполнение всех необходимых процедур до начала клинической оценки. План проведения исследований должен включать вопросы, отражающие выбор, оценку и характеристику методов, определение соответствующих точек отбора образцов, включая исходные образцы для определения предсуществующих антител, адекватные объемы проб, условия обработки (хранения) образцов, а также выбор статистических методов анализа полученных данных.
74. Эти положения относятся к методам, используемым для оценки и характеристики антител, и к методам, используемым для оценки клинических реакций, если установлено, что они вызваны антителами, индуцированными введением терапевтического белка. Многие из этих положений должны быть разработаны конкретно для каждого случая с учетом лекарственного препарата, пациентов и ожидаемых клинических проявлений.
6.5. Контрольные образы и реагенты
75. Идентификация и (или) разработка соответствующих положительных и отрицательных контролей имеет решающее значение, поскольку такие контроли необходимы для проведения валидации метода. Также использование положительных и отрицательных контролей тесно связано с интерпретацией результатов анализа и дифференциацией между серопозитивными и серонегативными образцами, то есть определением положительных и отрицательных образцов по наличию антител. Данные о характеристиках всех контролей, отражающих их свойства и адекватность возможности предполагаемого использования, должны быть представлены в регистрационном досье. Это особенно важно для положительного контроля, который во многих случаях является антителами животных, при этом необходимо учитывать, что их способность связываться с разными эпитопами может отличаться (например, в отношении препаратов биосимиляров).
76. Положительный контроль антител является образцом сыворотки, полученной от человека, которая содержит значительную концентрацию антител, объем сыворотки должен быть достаточным для дальнейшего использования. Однако достаточный объем сыворотки человека не всегда доступен, для того, чтобы использовать ее в качестве положительного контрольного образца. В таких случаях используются рекомбинантные антитела человека, специфичные к белку (при наличии), или используется в качестве референтного образца сыворотка животных, полученная при их иммунизации лекарственным препаратом. Эти требования также применимы и к биоподобным лекарственным препаратам. В связи с видовыми различиями использование антител животных имеет больше ограничений, чем антитела человека (например, при выполнении иммунохимических методик). Кроме того, если методика предусматривает использование реагента, специфичного к иммуноглобулину человека, он не будет адекватно взаимодействовать с антителами, иного происхождения (то есть с антителами, отличными от антител человека), и результаты таких исследований могут отличаться от результатов, полученных при определении антител человека, содержащихся в образцах плазмы человека.
77. В силу гетерогенности структуры, специфичности и авидности иммуноглобулинов, содержащихся в стандартах образцах и пробах, калибровка (градуировка) иммунологических методов является трудной задачей. Это обуславливает сложность или не невозможность, прямого сравнения исследуемых образцов и стандартных материалов (особенно по их массе). В связи с этим калибровку таких методов необходимо осуществлять, используя хорошо описанные и обоснованные подходы (например, необходимо оценить вариант представления данных иммуноферментного анализа в виде титра на основе стандартной процедуры расчета этой величины). При этом чувствительность методики и данные экспериментов, полученных методом добавок, должны быть представлены в количественных значениях. Положительные контроли антител для методов оценки нейтрализации должны обладать значительной нейтрализующей активностью, однако допускается включение в исследование препарата не-нейтрализующих антител, по крайней мере, в валидационных исследованиях, если такие антитела имеются. Нейтрализующую способность антител в каждом образце трудно определить в единицах массы и, поэтому определяют порог нейтрализующей активности антител. Такие пороговые значения (близкие к минимальному пределу обнаружения) должны быть надлежащим образом обоснованы и валидированы для обеспечения обнаружения всех положительных образцов с наличием нейтрализующих антител. Для учета результатов используются значения разбавления образца или титра, требуемого для нейтрализации биологической активности препарата.
78. Как для скрининговых методов, так и для методов определения нейтрализующей активности антител необходимо использовать набор стандартных материалов, содержащих различное количество антител (контроли низкого, среднего и высокого содержания антител), которые также используются для установления характеристик и валидации аналитических методик и которые служат показателями их пригодности. Набор стандартных материалов должен включать один или несколько препаратов с низким содержанием антител (близким к минимальному пределу обнаружения) и препаратов антител с низкой авидностью.
79. Отрицательные контроля необходимы для установления базовых показателей анализа, а также для характеристики и валидации методики. Исходные параметры аналитической методики у здоровых субъектов, как правило, достаточно легко определяются путем оценки результатов анализа образцов, полученных от достаточного количества таких субъектов, и обработки с целью получения статистически надежных исходных значений. Этот способ не всегда позволяет охарактеризовать исходные параметры аналитической методики при анализе образцов, полученных от пациентов, поэтому такие параметры определяются отдельно, с использованием образцов от пациентов, полученных до начала лечения, или образцов пациентов с тем же заболеванием, не участвующих в данном исследовании. Методы должны быть валидированы с использованием того же матрикса, что и анализируемые образцы.
80. В образцах некоторых субъектов исследования (пациентов) могут содержаться антитела, сформированные еще до начала лечения (предсуществующие антитела), или другие вещества, которые могут давать значимые ложноположительные результаты, поэтому для обеспечения правильности интерпретации результатов исследования образцов, полученных после лечения, в отношении выявления антител, индуцированных введением лекарственного препарата, необходим скрининг субъектов исследования (пациентов).
81. Реагенты, используемые при выполнении методик, должны быть квалифицированы (аттестованы) и установлены критерии приемлемости, по крайней мере, для тех, которые наиболее важны. Они должны быть охарактеризованы и храниться в надлежащих условиях (в лиофилизированном виде или замороженном состоянии при соответствующей температуре).
Валидация методов и интерпретация результатов
82. Методики, используемые для определения антител и нейтрализующих антител, содержащихся в образцах, полученных от пациентов, должны быть валидированы в отношении их соответствия, материалы по валидации должны быть включены в регистрационное досье. В то время как разработка и валидация методов являются непрекращающимся процессом на протяжении всей разработки препарата, данные по оценке иммуногенности в клинических исследованиях, которые включаются в регистрационное досье, должны быть получены с использованием валидированных методов. Валидационные исследования должны быть проведены для того, чтобы подтвердить, что используемые аналитические методики позволяют выявлять линейные, зависимые от концентрации ответы соответствующих аналитов, а также характеризуются соответствующей точностью, прецизионностью, чувствительностью, специфичностью и робастностью (устойчивостью, надежностью).
83. Необходимо обеспечить включение в валидационные документы данных в отношении предельных разведений образцов, позволяющих достоверно оценить результат. Во избежание межлабораторной вариабельности для выполнения анализов предпочтительным является использование централизованной лаборатории в рамках проведения клинических исследований, как предрегистрационных, так и после регистрации лекарственного препарата. Валидационные исследования должны быть проведены для подтверждения того, что эффект матрикса, обусловленный реактивами или веществами, содержащимися в образцах или интерференцией за счет присутствия лекарственного препарата, не влияет негативным образом на результаты выполненных исследований. Это осуществляется путем проведения исследований «восстановления» (recovery) и наблюдения за влиянием таких веществ, содержащихся в матриксе, на ответ, полученный в их отсутствие. Подобное исследование должно быть проведено в отношении всего диапазона разведений проб, используемых в анализах, и в некоторых случаях, по меньшей мере, в отношении предельных разведений, которые можно достоверно оценить.
84. Необходимо установить четкие критерии для определения параметров, позволяющих считать образцы положительными или отрицательными, а также условия подтверждения положительных результатов. Установленный подход должен быть обоснован полученными данными. Общим способом определения порога принятия результата как положительного при выполнении иммунологических методов является установление исходных параметров с использованием результатов оценки контрольных образцов здоровых лиц или пациентов. Для определения значения такого порога используются статистические методы. Для установления отсекаемых значений (cut-off) используется статистический подход если это обосновано. Допускается использование реальных данных (например, удвоенное значение, установленное при изучении исходных параметров) для определения результата, который будет принят в качестве минимального положительного значения. Для положительных образцов необходимо определить титр антител с использованием стандартного подхода и установить наибольшее разведение образца, при котором отмечается положительный результат. Альтернативным вариантом определения титра антител является проведение учета результатов в единицах массы, с использованием положительного контроля антител.
Такой подход имеет определенные ограничения (условия) для своего выполнения, указанные в пунктах 83 - 84 настоящей главы.
6.6. Методы оценки сравнительной иммуногенности
85. При разработке биоподобных лекарственных препаратов (биоаналогичных препаратов) всегда должны быть проведены сравнительные исследования иммуногенности, такие же исследования требуются при внесении определенных изменений в процесс производства биологического препарата. Указания по проведению таких исследований приведены в главах 9.1, 9.2 и 15 настоящих Правил.
86. Тестирование на иммуногенность биоподобного и референтного препаратов должно проводиться в рамках осуществления доказательства биоподобия с использованием одного и того же формата методов анализа (то есть должны быть использованы методики, базирующиеся на одинаковых принципах, и одинаковом режиме отбора образцов). Следует обеспечить, чтобы используемые методики обладали способностью выявлять антитела, направленные против всех эпитопов молекулы, как биоподобного так и референтного препаратов. Если для биоподобного и референтного препарата используются разные методики, требуется валидация методов для анализа двух антигенов в целях исключения любого влияния на результаты потенциальных различий методик в отношении их чувствительности и толерантности к лекарственному препарату. Демонстрация сходной частоты формирования антител и высокая степень соответствия между результатами анализа являются хорошим доказательством сопоставимости иммуногенности препаратов.
87. Заявитель вправе использовать одну методику, в которой биоподобная молекула используется в качестве антигена. Такой формат анализа позволяет обнаруживать все антитела к биоподобному лекарственному препарату, но не обязательно в таком случае выявляются все антитела, направленные к референтному препарату. Вывод о том, что биоподобный лекарственный препарат является более иммуногенным, должен инициировать исследование основной причины выявленных различий, включая методологические вопросы.
88. Независимо от подхода, используемого для оценки иммуногенности биоподобного лекарственного препарата по сравнению с референтным, методики должны быть перекрестно валидированы с использованием обоих антигенов, положительных контролей с наличием антител. В исследование необходимо включить клинические образцы для демонстрации сходства свойств препаратов.
89. Подходы к методологии анализа, указанные в пунктах 86 - 88 настоящей главы применимы и в тех случаях, когда требуются сравнительные исследования иммуногенности при оценке сопоставимости свойств 2 вариантов терапевтического белка, полученных до и после внесения изменений в процесс производства данного лекарственного препарата.
6.7. Оценка иммуногенности конъюгированных белков и слитых белков (fusion proteins)
90. В ответ на новые биотерапевтические молекулы, такие как сконструированные слитые белки (fusion proteins) и химически конъюгированные белки может наблюдаться формирование антител с различной специфичностью и вариабельной аффинностью к различным эпитопам, которые способны вызывать различные клинические последствия. Оценка такого гуморального ответа, в том числе характеристика специфичности индуцированных антител является сложной задачей и требует использования нескольких методик для определения иммунного ответа на различные фрагменты молекулы действующего вещества. Одним из вариантов анализа специфичности антител к индивидуальным фрагментам является использование стратегии, основанной на принципе конкурентного ингибирования при проведении подтверждающего анализа. Например, для пэгилированного белка в последовательность оценки его иммуногенности включается скрининговый анализ с использованием пэгилированного терапевтического препарата, затем в подтверждающем анализе при тестировании положительных образцов используется цельный терапевтический препарат, не-пэгилированный белок и фрагмент полиэтиленгликоля (ПЭГ).
6.8. Характеристика антител к терапевтическому белку
91. В стандартном случае требуется оценка частоты выявления и титров антител, длительности их персистенции и нейтрализующей способности. При определенных обстоятельствах, например, в случае развития анафилактоидных реакций, целесообразно провести дополнительную характеристику гуморального ответа, и проследить последующую динамику развития иммунного ответа. В таких случаях необходимо определение изотипа и подклассов IgG или функциональной активности Т-клеток. При подозрении на развитие аутоиммунных реакций необходимо изучить перекрестную реактивность индуцированных антител с соответствующими эндогенными белками.
7. Иммуногенность и клиническая разработка
92. Оценка иммуногенности должна быть частью клинических исследований фармакокинетики, фармакодинамики, безопасности и эффективности биологического лекарственного препарата, ориентированного на группы пациентов, которые ранее не подвергались воздействию препарата. Целью исследований иммуногенности является определение и характеристика иммунного ответа на препарат и исследование корреляций между формированием антител, с одной стороны, и параметров фармакокинетики, показателей фармакодинамики, а также эффективности и безопасности, с другой стороны. Таким образом, оценка иммуногенности должна быть включена в планирование основных клинических исследований, включая синхронизацию отбора образцов для определения антител и выбор соответствующих биомаркеров (при их наличии), также как оценка безопасности и эффективности. В описанном выше случае допускается не проводить специальные клинические исследования иммуногенности.
7.1. Обоснование режима отбора образцов и оценки кинетики формирования антител
93. Определение иммуногенности у пациентов должно проводиться систематически путем регулярного повторного отбора образцов. В случае развития симптомов, свидетельствующих о подозрении на развитие нежелательного иммунного ответа, проводят забор дополнительных образцов.
94. Отдельные факторы, связанные с лекарственным препаратом, потенциально влияют на развитие иммунного ответа на терапевтический белок. В связи с этим режим отбора образцов для обнаружения иммунного ответа необходимо адаптировать и подобрать индивидуально для каждого лекарственного препарата с учетом его характеристик, включая фармакокинетические свойства (например, период полувыведения) и влияние присутствия остаточного содержания лекарственного препарата в образцах на результаты анализа по выявлению антител (лекарственная толерантность методик, используемых для выявления антител). До начала терапии всегда должны быть взяты исходные образцы.
95. Заявители должны использовать общепринятую терминологию и подходы для описания кинетики гуморального ответа и потенциальных побочных реакций, обусловленных развитием нежелательного иммунного ответа, с учетом опыта применения подобных препаратов и терминологии, подходов используемых в научных руководствах и публикациях по иммунологии. Во время лечения образцы следует брать до очередного введения препарата, поскольку остаточное содержание действующего вещества в плазме может искажать результаты анализа.
96. Частота отбора образцов, а также сроки и объем проводимых анализов должны быть обоснованы, они зависят от степени риска иммуногенности, установленного в отношении конкретного лекарственного препарата (в соответствии с главой 11 настоящих Правил). Соблюдение режима отбора образцов позволяет выделить пациентов, у которых определяется временный, транзиторный положительный ответ, от пациентов, у которых формируется стойкий гуморальный ответ. Период отбора образцов после окончания лечения должен быть достаточно длительным для того, чтобы сделать вывод о стойкости иммунного ответа, вызванного терапевтическим белком, и выявить иммунные реакции, которые были подавлены самим терапевтическим белком. Сроки отбора образцов после окончания лечения определяются периодом полувыведения белка и лекарственной толерантностью методики по определению антител.
97. Более частый отбор образцов осуществляется на ранних этапах лечения, когда у пациентов наблюдается самый высокий риск выработки антител. Длительное наблюдение за иммуногенностью с менее частым отбором образцов дает дополнительную информацию о развитии и последствиях проявления иммуногенности. В отношении лекарственных препаратов, предназначенных для длительного непрерывного лечения, на предрегистрационном этапе необходимо получить данные по оценке иммуногенности в течение одного года. Данные за более короткий период наблюдения представляются только при наличии соответствующего обоснования.
98. Иммуногенность, ассоциированная с прерывистой схемой лечения, должна рассматриваться в отношении оценки риска с учетом опыта применения других аналогичных препаратов, рисков, связанных с потенциальной иммуногенностью, бустерного эффекта и сохранения или появления антител после воздействия препарата.
99. Если предусмотрены разные пути введения лекарственного препарата, на этапе подачи заявления о его регистрации заявитель должен обосновать подход к оценке иммуногенности при всех путях введения.
100. Риск побочных иммуно-опосредованных эффектов (побочных реакций), обусловленных проявлением нежелательной иммуногенности, должен быть кратко описан в соответствующих разделах инструкции по медицинскому применению лекарственного препарата (общей характеристике лекарственного препарата), с учетом того, что сравнение результатов, полученных из разных источников и (или) на основании использования разных методов анализа является ненадежным. Целесообразность и возможность регулярного мониторинга иммуногенности, включая необходимость определения концентрации лекарственного препарата, должны быть также включены в инструкцию по медицинскому применению лекарственного препарата, если это обосновано.
7.2. Влияние на фармакокинетику
101. Формирование антител может влиять на параметры фармакокинетики препарата, особенно на стадию его элиминации. Не-нейтрализующие, «связывающие» антитела иногда также могут изменять, а не просто снижать эффективность препарата (например, за счет увеличения периода полувыведения). Изменение параметров фармакокинетики может быть ранним признаком формирования антител. В связи с этим, заявителям необходимо включать во все исследования фармакокинетики повторного (многократного) введения лекарственного препарата дополнительный забор образцов для оценки, как фармакокинетических параметров, так и иммуногенности.
7.3. Влияние иммуногенности на безопасность и эффективность
102. Присутствие антител может иметь или не иметь клиническую значимость (то есть их наличие может вызывать или не вызывать клинические последствия). Крайне важно, чтобы клиническая разработка лекарственного препарата основывалась на анализе потенциальных рисков и возможности их обнаружения и смягчения клинических последствий. Планирование анализа иммуно-опосредованных побочных реакций должно основываться на анализе рисков, включая предыдущий опыт применения той же группы лекарственных препаратов, наличии потенциально иммуногенных структур в белке, а также популяции пациентов. У пациентов с предсуществующими антителами лекарственный препарат может проявлять отличающуюся эффективность и иметь иной профиль безопасности. Такие пациенты должны быть выделены в отдельную подгруппу (если это возможно), а их образцы проанализированы отдельно. При планировании исследований должны быть определены комплексы симптомов, которые могут быть ассоциированы с острой или замедленной гиперчувствительностью и аутоиммунной реактивностью, а также с потерей эффективности. Потенциальные иммунологические нежелательные реакции рассматриваются в плане управления рисками.
103. При установлении наличия антител необходимо, кроме титра и нейтрализующей способности, оценить дополнительные характеристики антител (например, класс иммуноглобулинов в случае развития реакций гиперчувствительности немедленного типа). Также необходимо провести дальнейшее изучение типичных клинически значимых антител, определить «пороговый» уровень антител, за пределами которого антитела оказывают существенное влияние на эффективность и (или) безопасность.
7.4. Методологические аспекты оценки сопоставимости иммуногенного потенциала как элемента сравнительных исследований
104. Прямые сравнительные исследования иммуногенности необходимы при разработке биоподобных лекарственных препаратов, а также в случае внесения изменений в процесс производства конкретного лекарственного препарата, когда проводят исследования по сопоставимости продуктов, полученных до и после внесения изменений (в соответствии с главами 9.1 и 9.2 настоящих Правил). При изменении процесса производства зарегистрированного лекарственного препарата требуется поэтапное проведение исследований сопоставимости. Если результаты первичных физико-химических и биологических исследований указывают на различия между лекарственными препаратами, полученными до и после внесения изменений в их производственный процесс, необходимо рассмотреть возможные последствия внесенных изменений на показатели безопасности и эффективности, включая влияние на иммуногенность. При необходимости проведения исследований иммуногенности вид и методика проведения этих исследований должны быть определены на основании анализа выявленных различий, пути введения, кривой зависимости «доза - ответ», терапевтического окна и возможного влияния на клиническую картину заболевания, а также опыта, ранее накопленного при разработке данного лекарственного препарата и для лекарственных препаратов этой группы. Оценку иммуногенности, как части клинического исследования при изучении сопоставимости, необходимо проводить путем прямого сравнения лекарственных препаратов, полученных до и после внесения изменений в процесс производства. В обоих случаях, при разработке биоподобных лекарственных препаратов и при внесении изменений в производственный процесс, целевая популяция при проведении клинических исследований безопасности, эффективности и иммуногенности должна быть чувствительной в отношении возможности выявления различий в иммуногенности препаратов и их влиянии на клинические показатели, а также быть репрезентативной относительно популяции, для которой лекарственный препарат предназначен. В случае высокого риска применения лекарственного препарата образцы должны анализироваться на постоянной основе (на протяжении всего клинического исследования).
105. Исследование иммуногенности и оценка их результатов должны проводиться в комплексе с исследованиями фармакокинетики, безопасности и эффективности.
106. Различия в иммуногенности будут подвергать сомнению сопоставимость биоподобного и референтного препаратов, а также новых и старых версий ранее зарегистрированного препарата, кроме того, потребуется тщательный анализ основных причин выявленных различий. Минимальные различия в иммуногенности без корреляции на качественном уровне и без отрицательного воздействия на клиническую эффективность (снижение или потеря эффективности) и безопасность могут быть приемлемыми. Оценка клинической значимости наблюдаемых различий в иммуногенности может вызвать определенные сложности из-за ограниченного размера выборки и продолжительности наблюдения. Если клиническая значимость наблюдаемых различий является неопределенной (например, из-за низкой частоты потенциально серьезных неблагоприятных воздействий или медленного развития иммунного ответа), может потребоваться специальная стратегия управления рисками и обновление плана управления рисками (как описано в главе 9 настоящих Правил).
7.5. Управление иммуногенностью
107. Развитие нежелательных иммунных реакций на терапевтический белок нельзя полностью исключить, даже в случае выбора производителем в качестве целевого соединения - соединения с низким иммуногенным потенциалом. В связи с этим заявитель должен изучить возможность снижения неблагоприятного воздействия иммуногенности, наблюдаемого на этапе клинической разработки препарата (если это применимо).
108. В некоторых случаях применение иммуносупрессивных или противовоспалительных лекарственных препаратов в качестве сопутствующей терапии может значительно предотвращать или снижать иммуно-опосредованные нежелательные реакции.
109. Например, при применении препаратов коагуляционных факторов, может быть предпринята попытка восстановления иммунологической толерантности к препарату посредством использования толерогенных схем (например, путем введения больших доз терапевтического белка). Такие терапевтические схемы должны быть обоснованы результатами клинических исследований и отражены в соответствующих документах. Заявитель должен включить в инструкцию по медицинскому применению лекарственного препарата рекомендации для лечащего врача о том, каким образом можно смягчить последствия проявлений иммуногенности препарата.
8. Фармаконадзор
110. В состав регистрационного досье должен быть включен план управления рисками в соответствии с актами органов Союза в сфере обращения лекарственных средств и Правилами практики фармаконадзора. Вопросы иммуногенности должны быть отражены в разделе спецификации безопасности плана управления рисками терапевтических белков, и в случае необходимости должен быть рассмотрен вопрос о проведении дополнительных мероприятий в рамках фармаконадзора. При внесении изменений в производственный процесс, оценку влияния этого изменения на иммуногенный потенциал, необходимо предусмотреть в плане управления рисками. Оценка иммуногенности требует междисциплинарного подхода и для обеспечения лучшего решения вопросов необходимо обязательное совместное участие в исследованиях специалистов по качеству, доклиническим и клиническим исследованиям.
111. Объем данных по иммуногенности, полученных в ходе программы клинической разработки терапевтического белка до его регистрации, зависит от частоты развития иммунного ответа и связанных с ним рисков, обусловленных как иммуногенным потенциалом белка в популяции (популяциях), получающих данный препарат, так и частотой встречаемости заболевания. В связи с этим, наличие данных об иммуногенности на момент регистрации препарата может быть ограничено. Сведения полученные для данной группы препаратов и (или) референтного препарата (в случае разработки биоподобного (биоаналогичного) лекарственного препарата), должны быть представлены в полном объеме в соответствующих разделах резюме по клинической безопасности. На основе всех имеющихся данных в резюме по клинической безопасности делается заключение о том, представляет исследуемый препарат значимый (потенциальный) риск в отношении иммуногенности или не представляет.
112. В случае если лекарственный препарат представляет значимый (потенциальный) риск в отношении иммуногенности, иммуногенность должна быть отражена в плане управления рисками как потенциальный или идентифицированный риск или как раздел, требующий получения дополнительной информации. Иммуногенность препарата всегда связана с клинической значимостью. Если итоговая оценка иммуногенности не определяется как «сомнительная» или «неопределенная», то включение исследований иммуногенности, в отношении определения потенциального риска или получения недостающей информации, не требуется.
113. В рамках плана фармаконадзора в плане управления рисками должна быть указана необходимость проведения дополнительных мероприятий фармаконадзора. В случае если проведение дополнительных исследований иммуногенности необходимо, должен быть разработан наиболее подходящий дизайн в соответствии с целью исследования. В настоящее время выявление антител и определение минимального уровня антител не являются рутинными в клинической практике. Чтобы получить дополнительные данные по частоте выявления, титрам и минимальному уровню антител, допускается проведение дополнительных клинических исследования или продолжение текущих клинических исследований в пострегистрационном периоде. Такие клинические исследования также допускается проводить при разработке биоподобного (биоаналогичного) лекарственного препарата, если дополнительные данные по иммуногенности необходимо получить при проведении сравнительных исследований в пострегистрационном периоде.
114. Последующее наблюдение за пациентами, получавшими терапевтический белок во время обычной клинической практики, (например, с помощью реестров пациентов), а также сбор спонтанно сообщаемых подозрительных побочных реакций показал, что указанные мероприятия позволяют обеспечить необходимый сбор данных о безопасности таких препаратов. Указанные мероприятия фармаконадзора необходимо также использовать для идентификации нежелательных реакций, обусловленных иммуногенностью, таких как реакция, связанная с инфузией, или истинная красноклеточная аплазия при применении эритропоэтинов. Основываясь на полученных данных, необходимо сделать заключение о потенциальном нежелательном иммунном ответе, с учетом наличия признаков и (или) симптомов, указывающих на нарушение безопасности и (или) потерю эффективности лекарственного препарата, включая изменения соответствующих биомаркеров. Все эти вопросы должны быть отражены в плане управления рисками.
115. Если иммуногенность включена в раздел технических требований безопасности плана управления рисками, должна быть проанализирована целесообразность проведения дополнительных мероприятий с целью минимизации рисков. В этом случае все указанные дополнительные мероприятия должны быть четко описаны. Рутинные действия с целью минимизации рисков, связанных с иммуногенностью, также должны быть отражены в инструкции по медицинскому применению лекарственного препарата, включая сведения о том, как выявлять и определять минимальный уровень антител, предотвращать формирование антител и связанные с ними нежелательные реакции.
116. Для терапевтических белков критически важно решить все вопросы, связанные с идентификацией препарата и его номера серии, которая предположительно вызвала нежелательную реакцию, а также прослеживаемость на фармацевтическом рынке. Это особенно важно для нежелательных реакций, связанных с проявлением иммуногенности, независимо от того, были ли они выявлены в рамках обычного фармаконадзора и (или) дополнительных мероприятий по фармаконадзору. Кроме того, должны быть приняты меры по оптимизации прослеживаемое™ препарата на фармацевтическом рынке, в частности, сбора сведений о торговом наименовании лекарственного препарата и номере серии.
9. Резюме программы иммуногенности
117. Как планирование, так и оценка исследований иммуногенности биологического лекарственного препарата требует междисциплинарного подхода. Данные, имеющие отношение к оценке иммуногенности, распределяются по различным разделам регистрационного досье, представляемых для регистрации препарата. В связи с этим заявитель включает в регистрационное досье комплексное резюме по иммуногенности, в том числе обоснование выбранной программы изучения иммуногенности. Краткое резюме программы изучения иммуногенности размещают в разделе 2.7.2.4 модуля 2 регистрационного досье, а более подробные данные в разделе 5.3.5.3 модуля 5 регистрационного досье. Резюме должно быть кратким и содержать ссылки на соответствующие главы настоящих Правил и приложения к ним.
118. Резюме с оценкой риска должно отражать исследования на протяжении всего жизненного цикла лекарственного препарата и должно быть использовано для обоснования исследований на различных этапах разработки препарата.
119. Оценка риска может свидетельствовать о низком риске побочных реакций, обусловленных проявлением нежелательной иммуногенности. Тем не менее иммуногенность должна изучаться с помощью использования валидированных методов в соответствии с приложением к настоящей главе. Отклонение от этой схемы (например, отсутствие тестирования на нейтрализующие свойства антител при проведении клинических исследований однократных доз терапевтических белков с низким риском), должно быть обосновано. На основании оценки рисков необходимо решить вопрос о проведении изучения дополнительных характеристик иммунного ответа (например, изотипирование антител и картирование эпитопов), а также определяется частота отбора образцов, время проведения анализа и выбор целевой популяции.
120. Резюме программы иммуногенности включает в себя следующие разделы (если применимо):
а) анализ факторов риска;
б) программу оценки риска, связанного с иммуногенностью;
в) результаты оценки иммуногенности;
г) выводы о риске (рисках), связанных с иммуногенностью.
121. Раздел резюме программы иммуногенности «Анализ факторов риска» включает в себя следующие подразделы:
а) предыдущий опыт применения лекарственного препарата (лекарственных препаратов данной группы):
наличие у лекарственного препарата эндогенного аналога;
наличие моделей животных, которые позволяют оценить потенциальные последствия иммуногенности (например, элиминация эндогенного белка);
наличие сведений об антигенных участках молекулы;
предпринятые производителем попытки снизить проявления иммуногенности препарата до и во время проведения клинических исследований;
б) физико-химические и структурные характеристики лекарственного препарата:
наличие в молекуле действующего вещества лекарственного препарата новых потенциально иммуногенных структур (например, последовательностей, чужеродных для организма человека);
экспрессирующая конструкция и посттрансляционный профиль (например, отличный от человека профиль гликозилирования (гликаны в составе молекулы лекарственного препарата));
стабильность и примеси (например, наличие агрегатов (видимые и невидимые частицы));
состав (производственная рецептура) и система упаковки (укупорки) лекарственного препарата (например, потенциальные примеси и возможность выщелачивания);
в) потребность в особых мерах фармаконадзора в связи с настороженностью (беспокойством) связанным с предлагаемым путем введения и (или) способом введения лекарственного препарата;
г) оценка факторов, связанных с пациентом или обусловленных природой (патогенезом) заболевания:
статус иммунологической толерантности (в том числе: склонность к аутоиммунным реакциям, отсутствие иммунологической толерантности (например, дефекты генов, кодирующих эндогенные белки), сопутствующая иммуномодулирующая терапия);
предсуществующий у пациента иммунитет (в том числе «естественные» антитела, антитела, индуцированные ранее проведенной терапией препаратами, содержащими родственные субстанции).
122. Раздел резюме программы иммуногенности «Программа оценки риска, связанного с иммуногенностью» должен включать в себя следующие подразделы:
а) стратегия анализа или методология исследования:
обоснование выбора методов анализа (в том числе: скрининг, подтверждение положительных результатов, определение титров антител, определение их нейтрализующей активности, другие характеристики антител (например, определение класса и подкласса иммуноглобулинов));
специфичность и чувствительность выбранных методов для конкретного лекарственного препарата (в том числе: выбор положительного контроля (контролей), определение порогового значения, свидетельствующего о наличии антител (то есть порогового значения для сероположительных образцов));
устойчивость методики количественного анализа лекарственного препарата к влиянию других мишеней (отсутствие влияния остаточного содержания в образце лекарственного препарата других антигенов-мишеней на чувствительность методики);
эффект матрикса в различных популяциях (искажение результатов за счет интерференции матрикса в различных популяциях);
б) подход к оценке иммуногенности при проведении клинических исследований (дизайн оценки):
отбор образцов для тестирования на иммуногенность; обоснование продолжительности наблюдения (в том числе в период проведения терапии, без терапии, после окончания терапии);
фармакокинетика (в том числе: возможное искажение результатов анализа по выявлению антител (интерференция) за счет определенной концентрации лекарственного препарата, пороговый уровень содержания лекарственного препарата в отношении толерантности методики по выявлению антител к его присутствию в тестируемом образце (или пороговый уровень лекарственного препарата в отношении фармакологической интерференции на результаты анализа по выявлению антител));
исследование фармакодинамики, эффективности и безопасности (в том числе: выявление частоты, продолжительности и (или) стойкости выявления и клинической значимости потенциальных антител, реакции гиперчувствительности, аутоиммунные реакции, снижение эффективности лекарственного препарата, выявление типа гуморального ответа с формированием антител (стойкий, транзиторный) и потенциально иммуноопосредованных отдельных симптомов и синдромов в соответствии с определениями и терминологией, принятой в научной литературе (публикациях) по иммунологии, оценка корреляции клинических последствий с наличием антител);
в) влияние оценки риска на программу изучения иммуногенности.
123. Раздел резюме программы иммуногенности «Результаты оценки иммуногенности» должен включать в себя следующие подразделы:
а) оценка иммуногенности в клинических исследованиях (оценка сравнительной иммуногенности в случае внесения изменений в процесс производства и при разработке биоподобного лекарственного препарата):
частота выявления (сравнительная оценка) антител, включая нейтрализующие антитела;
титры и продолжительность (стойкость) выявления антител (сравнительная оценка);
развернутая характеристика антител, если она необходима (например, определение класса иммуноглобулинов, перекрестная реактивность с соответствующими терапевтическими или эндогенными белками);
б) воздействие антител (сравнительная оценка) на фармакокинетику, фармакодинамику, безопасность и эффективность лекарственного препарата;
в) воздействие предсуществующих антител на фармакокинетику, фармакодинамику, безопасность и эффективность лекарственного препарата.
124. Раздел резюме программы иммуногенности «Выводы о риске (рисках), связанных с иммуногенностью» должен включать в себя следующие подразделы:
а) влияние иммуногенности на соотношение «польза - риск»;
б) подходы по управлению рисками (вопросы управления рисками):
идентификация групп риска;
наличие безопасного уровня или типа проявления иммуногенности;
необходимость премедикации, сопутствующей терапии;
возможность деиммунизации (терапия индукции иммунологической толерантности);
способы выявления и смягчения (минимизация) рисков;
в) оценка взаимосвязи нежелательных реакций с проявлением иммуногенности в пострегистрационном периоде (план управления рисками).
Приложение
к главе 25 Правил проведения
исследований биологических
лекарственных средств Евразийского
экономического союза
ПРИМЕР
стратегии (методологии) оценки иммуногенности
Таблица 1
Часто используемые скрининговые методы
| Методы (тип анализа) | Преимущества (достоинства) | Недостатки |
|---|---|---|
| Прямой или непрямой иммуноферментный анализ (метод ELISA) | высокая производительность недорогой простота в использовании и высокая степень автоматизации высокая толерантность к белку препарата в жидкой фазе доступность реагентов и оборудования | возможно неспецифическое связывание потенциально высокие фоновые значения иммобилизация антигена может изменять конформацию антигена и привести к маскировке эпитопов могут не выявляться низкоаффинные антитела низкая толерантность к белку лекарственного препарата при твердофазном варианте требуются видоспецифичные вторичные реагенты |
| Модифицированный иммуноферментный анализ (Bridging ELISA) | высокая производительность недорогой простота в использовании и высокая степень автоматизации низкий уровень фоновых значений высокая специфичность (за счет двойного связывания) могут быть использованы межвидовые перекрестно-реагирующие реагенты доступность реагентов и оборудования | метка антигена может изменять его свойства низкоаффинные антитела могут не выявляться высокая чувствительность к интерференции терапевтическим препаратом, сывороточными компонентами (например, антитела к Ig человека, мультивалентные мишени) могут не выявляться антитела классов IgG4 и IgM |
| Электрохемилюминесценция (с прямым и (или) непрямым связыванием) | высокая производительность большой динамический диапазон минимальное влияние матрицы высокая толерантность к белку препарата сигнал обнаружения постоянный в период сохранения активности конъюгата TAG | возникает потребность в двух конъюгатах антигена (в непрямом тесте) метка антигена может изменять его свойства чувствительность к интерференции терапевтическим препаратом, сывороточными компонентами (например, антитела к Ig человека, мультивалентные мишени) могут не выявлять антитела классов IgG4 требуется специальное оборудование и реагенты |
| Метод радиоиммунопреципитации | средняя производительность высокая чувствительность может быть строго специфичным недорогой | может быть изотип специфичным могут не выявляться низкоаффинные антитела требует использования радиоактивномеченного антигена распад радиометки может влиять на стабильность антигена |
| Поверхностный плазмонный резонанс | средняя производительность определяет специфичность, изотип, относительную аффинность связывания выявляет низкоаффинные и высокоаффинные антитела высокая - высокая толерантность к белку препарата не требуются реагенты для выявления антител | иммобилизация антигена может изменить терапевтический белок стадия регенерации может вызвать деградацию антигена чувствительность может быть ниже, по сравнению с методиками связывания высокая стоимость требуется специальное оборудование и реагенты |
Таблица 2
Методы определения нейтрализующей активности антител
| Методы (тип анализа) | Преимущества (достоинства) | Недостатки |
|---|---|---|
| Биологические методы с использованием клеточных культур | определяет функциональные свойства, отражающие механизм действия препарата результаты как правило коррелируют с клиническим ответом | относительно трудоемкий - возможен сложный дизайн высокая частота вариабельности подвержен влиянию сывороточных матриксных эффектов и других факторов интерференции чувствительность к интерференции за счет препарата валидация может быть трудновыполнимой, за счет использования клеточных линий, реагентов и др. |
| Метод конкурентного связывания лигандов | быстрый простой дизайн относительно легко выполнимый не требует использования клеточных культур простота разработки и валидации | метка антигена может изменить его свойства чувствительность к интерференции за счет препарата не позволяет оценить истинную функциональную активность может не коррелировать с клиническим ответом |
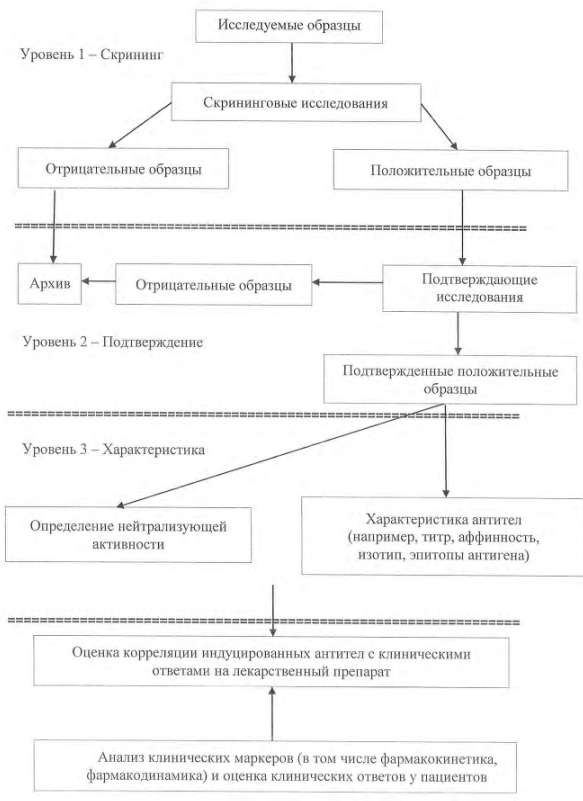
Рисунок. Пример стратегии оценки иммуногенности
Глава 26. Указания по проведению клинических исследований рекомбинантных и полученных из плазмы крови человека лекарственных препаратов фактора IX
1. Общие положения
1. Настоящая глава содержит указания по формированию материалов раздела клинической документации, которые должны быть включены в регистрационное досье, представляемое для регистрации лекарственных препаратов фактора свертывания крови IX, полученных на основе технологии рекомбинантных ДНК или из плазмы крови человека, предназначенных для лечения и профилактики кровотечений у пациентов с гемофилией В (далее - препараты фактора IX) и по правилам проведения клинических исследований препаратов фактора IX до их регистрации и в пострегистрационном периоде. Отдельно рассматриваются вопросы, связанные с внесением значительных изменений в производственный процесс ранее зарегистрированных препаратов фактора IX.
2. Целью настоящей главы, является формирование гармонизированных требований к материалам клинических исследований, представляемым для регистрации препаратов фактора IX.
3. Сравнение фармакокинетических параметров рекомбинантного фактора IX и фактора IX, получаемого из плазмы крови человека, показало, что периоды полувыведения почти идентичны, тогда как восстановление in vivo статистически различалось. Низкое восстановление рекомбинантного фактора IX по сравнению с плазменным фактором IX связано с различием в сульфатировании и отсутствием фосфорилирования рекомбинантного фактора IX.
4. В материалы регистрационного досье, должны быть включены данные клинических исследований по оценке безопасности и эффективности с точки зрения иммуногенности и других нежелательных реакций, проведенных с участием пациентов всех возрастных групп. В зависимости от типа препарата фактора IX необходимо провести исследования на не получавших ранее лечение пациентах, чтобы изучить безопасность и эффективность у этой особой популяции пациентов. В отношении препаратов фактора IX необходимо изучить тромбогенный потенциал лекарственного препарата.
5. Приведенные в настоящей главе указания по проведению клинических исследований, необходимых для регистрации препаратов фактора IX относятся к:
лекарственным препаратам, заявленным на регистрацию в качестве новых лекарственных препаратов;
ранее зарегистрированным лекарственным препаратам, в производственный процесс которых внесены значительные изменения (например, дополнительные стадии инактивации и (или) элиминации вирусов или новые способы очистки).
6. Клинические исследования, описанные в настоящей главе, должны выполняться в соответствии с требованиями Правил надлежащей клинической практики Евразийского экономического союза.
7. Общие принципы оценки безопасности и эффективности при проведении клинических исследований отражены в разделах 2 и 3 настоящей главы. Информация об особенностях клинической разработки новых лекарственных препаратов и ранее зарегистрированных лекарственных препаратов, в производственный процесс которых внесены значительные изменения, включена в последующие разделы настоящей главы.
8. Для препаратов фактора IX, имеющих определенные особенности (например, длительный период полувыведения), требуется модификация клинического исследования, дизайн клинических исследований в данном случае необходимо предварительно согласовать с уполномоченным органом (экспертной организацией) государства-члена.
9. При проведении клинических исследований необходимо учитывать, что фармакокинетика препаратов фактора IX, рекомбинантного и лекарственного препарата, полученного из плазмы крови, отличается. Результаты сравнительных исследований фармакокинетических параметров показали, что периоды полувыведения фактора IX, рекомбинантного и лекарственного препарата, полученного из плазмы крови, практически идентичны, тогда как показатели восстановления активности фактора IX in vivo после инфузии лекарственных препаратов статистически значимо различаются. Объяснением наблюдающегося более низкого уровня восстановления активности рекомбинантного фактора IX in vivo по сравнению с фактором IX, полученным из плазмы крови, служит отсутствие фосфорилирования остатка Ser158 и низкий уровень сульфатирования Tyr155 в рекомбинантном факторе IX.
10. В разделе 6 настоящей главы приведены указания по проведению клинических исследований с участием ранее не получавших лечение пациентов для оценки безопасности и эффективности препаратов фактора IX при лечении этой конкретной популяции пациентов. Условия проведения данных исследований зависят от типа исследуемого препарата фактора IX.
11. Детальные требования к клиническим исследованиям препаратов фактора IX представлены в приложениях № 1 - 3 к настоящей главе.
12. Положения настоящей главы касаются вопросов клинических исследований, которые должны проводиться до и после регистрации препаратов фактора IX. Вопросы, касающиеся оценки качества препаратов фактора IX, не рассматриваются в настоящей главе.
13. При проведении исследований в детской популяции заявитель клинического исследования должен также выполнять требования актов органов Союза в сфере обращения лекарственных средств и законодательства государств-членов к организации исследований в педиатрической практике.
2. Оценка эффективности
Неэффективность препаратов фактора IX используемых для лечения пациентов с гемофилией В, необходимо подтвердить в клинических исследованиях, которые должны быть проведены до их регистрации. Обязательным условием является проведение последующих пострегистрационных исследований для сбора дополнительных клинических данных и обеспечения согласованности в долгосрочной перспективе результатов предрегистрационных клинических исследований и рутинного применения препаратов фактора IX.
15. При клинической оценке препаратов фактора IX, изначально исследуют фармакокинетику основного действующего фактора. Для оценки фармакокинетических параметров нового препарата фактора IX наиболее важными являются следующие суррогатные конечные точки:
показатель восстановления активности фактора;
период полувыведения, площадь под кривой (AUC);
клиренс.
16. Оценка эффективности лечения препаратом фактора IX складывается из оценки профилактической эффективности - при регулярном использовании препарата для профилактики спонтанных кровотечений, а также терапевтической эффективности - при его использовании по требованию для купирования уже развившихся кровотечений. Оценка проводится, как самими пациентами, так и лечащим врачом за период, как минимум, 50-дневного введения препарата.
3. Оценка безопасности
17. Вопросы безопасности препаратов фактора IX включают в себя оценку вирусной безопасности, иммуногенности и других нежелательных реакций. Использование при производстве рекомбинантных препаратов нечеловеческих клеточных линий повышает вероятность присутствия в них различных контаминантов. Кроме того, потенциальное присутствие гетерологичных белков повышает иммуногенный потенциал таких лекарственных препаратов. Возможно развитие реакций гиперчувствительности к гетерологичным белкам (например, к белкам мыши, крупного рогатого скота или хомячков). Тромбогенность (риск развития тромбообразования) также должна рассматриваться как потенциальная проблема безопасности.
4. Нежелательные реакции
18. Во время проведения клинических исследований у всех пациентов, получающих препарат фактора IX, должны оцениваться параметры безопасности, включая оценку влияния лекарственного препарата на жизненно важные показатели. Все нежелательные реакции, проявившиеся при клинических исследованиях, должны быть зарегистрированы и проанализированы в отношении причины их возникновения, тяжести проявления и ожидаемости.
19. Все нежелательные реакции, связанные с любым применением препарата фактора IX, подлежат регистрации, а информация о них должна быть передана уполномоченному органу государства-члена в соответствии с требованиями Правил практики фармаконадзора.
20. В зависимости от типа препарата фактора IX, развитие реакций гиперчувствительности к гетерологичным белкам (например, к белкам мыши, крупного рогатого скота или хомячков) может проявляться в виде соответствующих нежелательных реакций, которые необходимо регистрировать. Все протоколы исследований должны включать опросный лист (регистрационную форму) для сбора соответствующих данных по реакциям гиперчувствительности.
5. Безопасность в отношении вирусов и других трансмиссивных агентов
5.1. Рекомбинантные лекарственные препараты
21. Безопасность рекомбинантных лекарственных препаратов в отношении вирусной контаминации обеспечивается путем тестирования вирусов в процессе производства и внедрения в производственный процесс стадий инактивации и (или) элиминации вирусов в соответствии с главой 2 настоящих Правил.
5.2. Лекарственные препараты фактора IX, полученные из плазмы крови человека
22. Производители должны обеспечить вирусную безопасность лекарственных препаратов, получаемых из плазмы крови человека, включая препараты фактора IX, путем отбора доноров, тестирования индивидуальных донаций и пулов плазмы крови на специфичные маркеры инфекций, а также путем включения в процесс производства эффективных стадий инактивации и (или) элиминации вирусов. Аналогичные принципы, относящиеся к обеспечению вирусной безопасности, должны применяться в отношении всех трансмиссивных агентов, включая агентов губчатой энцефалопатии и другие потенциальные патогены. Требования по обеспечению вирусной безопасности приведенные в главах 2 и 3 настоящих Правил, распространяются на данную группу лекарственных препаратов.
23. Используемые в производстве способы инактивации и (или) элиминации вирусов указанные в главах 2 и 3 настоящих Правил в настоящее время считаются высокоэффективными и обеспечивают вирусную безопасность лекарственных препаратов в отношении широкого спектра оболочечных вирусов. В связи с этим при проведении клинических исследований оценивать вирусную безопасность лекарственных препаратов в отношении оболочечных вирусов не требуется.
24. Используемые в процессе производства способы инактивации и (или) элиминации вирусов указанные в главах 2 - 3 Правил имеют определенные ограничения в отношении безоболочечных вирусов, таких как вирус гепатита А и парвовирус В19. При проведении клинических исследований в настоящее время не может быть адекватно оценена безопасность лекарственных препаратов, полученных из плазмы крови человека в отношении безоболочечных вирусов.
25. Заявитель должен представить все имеющиеся данные о пациентах, прошедших лечение лекарственным препаратом, полученным из плазмы крови человека в клинических исследованиях.
26. После окончания исследования за пациентами должны быть продолжены наблюдения в соответствии с обычной клинической практикой. Должна быть разработана информационная система, содержащая сведения о рисках для безопасности и описание мер по составлению отчетов о нежелательных реакциях. Заявитель также должен подтвердить, что разработана и функционирует система сбора информации о пациентах, принимавших лекарственный препарат, полученный из плазмы крови человека, которая позволяет быстро реагировать на любые сообщения о заражении пациента с последующим полным расследованием его причины.
5.3. Иммуногенность лекарственных препаратов фактора IX, полученных из плазмы крови человека
27. Иммуногенность препаратов фактора IX должна быть исследована до его регистрации и подтверждена результатами исследования в пострегистрационном периоде.
28. Заболевание гемофилией В наблюдается примерно в 4 раза реже, чем гемофилией А. Частота формирования ингибиторов у пациентов с гемофилией В после введения фактора IX ниже по сравнению с частотой выявления ингибиторов у пациентов с гемофилией А. Ингибиторы фактора IX выявляются приблизительно у 4% пациентов с тяжелой формой гемофилии В. Установлено, что формирование ингибиторов обычно ассоциируется с полной делецией гена фактора IX. Оценка иммуногенности препаратов фактора IX, где это применимо, проводится на основе тех же принципов, которые применяются для проведения клинических исследований препарата фактора VIII свертывания крови для лечения гемофилии А.
29. В отличие от гемофилии А, у пациентов с гемофилией В чаще развиваются анафилактические реакции на препараты фактора IX, ассоциированные с формированием ингибиторов. Образование антител, нейтрализующих фактор IX, снижает эффективность лечения, что требует постоянного увеличения количества вводимых доз фактора IX или введения очень больших доз препарата для индукции иммунологической толерантности. В научной медицинской литературе имеется информация о развитии анафилактических реакций, а также нефротического синдрома при терапии, проводимой с целью формирования иммунологической толерантности к препарату. Указанные проблемы касаются как препаратов, получаемых из плазмы, так и препаратов рекомбинантного фактора IX.
30. У пациентов с развитием анафилактических реакций, или у которых выявлены ингибиторы к фактору IX, необходимо путем использования соответствующих методов провести определение специфических иммуноглобулинов класса Е или G (IgE, IgG) к фактору IX.
5.4. Тромбогенность лекарственных препаратов фактора IX
31. Лечение препаратами фактора IX, полученными из плазмы, которые содержат факторы II, VII и X, может вызывать тромбозы. Препараты фактора IX, которые характеризуются более высокой степенью очистки, демонстрируют меньший риск развития тромбоэмболических осложнений. Клинические исследования новых препаратов фактора IX должны включать в себя определение маркеров активации коагуляции (фрагментов протромбина 1 + 2, комплексов тромбин-антитромбин (ТАТ) и D-димеров) с помощью использования соответствующих тестов в образцах, взятых у пациентов до и после инфузии, в период отсутствия кровотечения. Указанные исследования должны быть выполнены у пациентов, участвующих в фармакокинетическом исследовании. Клиническая оценка риска развития тромбозов должна проводиться безопасными, объективными способами, как минимум, у 5 пациентов, которым потребовалось не менее 10 хирургических вмешательств.
6. Представление документов о клинических исследованиях на регистрацию лекарственных препаратов фактора IX, заявляемых как новые лекарственные препараты
32. Положения настоящего раздела применяются к рекомбинантным и плазменным препаратам фактора IX, заявленным на регистрацию.
6.1. Общие положения, касающиеся клинических исследований
33. Принимая во внимание, что гемофилиия В относится к орфанным заболеваниям, результатов предрегистрационных исследований недостаточно для оценки всех аспектов терапии препаратами фактора IX, особенно безопасности, связанной с иммуногенностью.
34. В связи с этим для сбора дополнительных клинических данных и обеспечения согласованности в долгосрочной перспективе результатов предрегистрационных клинических исследований и рутинного применения должны быть проведены пострегистрационные исследования препарата фактора IX.
35. В предрегистрационные клинические исследования, должно быть включено не менее 40 пациентов. Указанное количество пациентов является оптимальным по соотношению необходимых клинических данных для оценки безопасности и эффективности препарата фактора IX, и доступностью пациентов, страдающих орфанным заболеванием. Ожидается, что указанное количество пациентов будет достаточно для получения достоверной информации об общих аспектах безопасности и демонстрации эффективности клинического применения препарата фактора IX с точки зрения его способности восстановить уровень фактора IX и достичь гемостаза, купировать развившиеся, а также предотвратить спонтанные кровотечения.
36. Учитывая, что в предрегистрационных исследованиях принимает участие ограниченное количество пациентов, дополнительная информация, в основном касающаяся аспектов безопасности, должна быть получена при проведении пострегистрационных исследований.
37. Клиническая разработка препаратов фактора IX должна основываться на поэтапном подходе в выборе пациентов, участвующих в клинических исследованиях, чтобы обеспечить возможность клинической оценки препарата на взрослых пациентах и детях старшего возраста, прежде чем в исследования будут включены дети младшего возраста. Изначальной возрастной когортой, подлежащей исследованию, являются ранее получившие лечение пациенты в возрасте 12 лет и старше. После того, как фармакокинетика, безопасность и эффективность будут оценены у 10 ранее получивших лечение пациентов 12 лет и старше получивших, по меньшей мере 50-дневное введение препарата, возможно инициирование проведения клинического исследования с участием детей в возрасте от 0 до 12 лет. Клинические исследования детей в возрасте от 0 до 12 лет должны начинаться с изучения фармакокинетики с последующим исследованием безопасности и эффективности, по меньшей мере, 50-дневного введения препарата у каждого из 20 пациентов. Результаты оценки фармакокинетических параметров, профиля безопасности и эффективности должны быть получены в рамках предрегистрационных исследований препарата фактора IX.
38. При проведении исследований в детской популяции заявитель клинического исследования должен также выполнять требования актов органов Союза в сфере обращения лекарственных средств и законодательства государств-членов к организации исследований в педиатрической практике.
39. Клинические исследования с участием ранее не получавших лечение пациентов должны проводиться при разработке всех новых лекарственных препаратов на основе рекомбинантного фактора IX (препаратов на основе новых генетических конструкций или модификаций молекулы фактора IX, выполненных с целью изменения его свойств in vivo (например, параметров фармакокинетики). Подобные исследования также проводятся для препаратов фактора IX, изготовленных с использованием новых способов получения рекомбинантного белка (например, новой линии клеток), имеющей ограниченный опыт применения.
40. Отсутствие данных в отношении ранее не получивших лечение пациентов необходимо указать в разделе 4.2 общей характеристики лекарственного препарата. Дозировка и способ применения не включаются в инструкцию по медицинскому применению препарата фактора IX до тех пор, пока не будут представлены результаты оценки безопасности и эффективности по 20 ранее не получившим лечение пациентам, получившим, как минимум 50-дневное введение препарата.
41. В случае препаратов фактора IX, полученных из плазмы (например, с использованием новых способов производства), потребность в исследованиях с участием ранее не получивших лечение пациентов рассматривается в каждом конкретном случае отдельно.
42. Вопросы выбора дизайна исследования и оцениваемых параметров в зависимости от популяции и целей исследования приведены в приложениях № 1 и 2 к настоящей главе.
6.2. Определение активности препарата фактора IX
43. В связи с наличием нескольких методик, используемых для определения фактора IX, значения оцениваемых показателей активности препарата могут значительно отличаться в зависимости от используемого метода, реагентов и стандартных образцов. Эти несоответствия, связанные с используемым методом, могут влиять как на маркировку готового лекарственного препарата, так и на результаты мониторинга пост-инфузионных образцов.
44. Указание активности фактора IX должно быть проведено в соответствии с методикой предложенной в «Рекомендациях по указанию активности концентратов факторов VIII и IX» Международного общества тромбоза и гесмотаза (ISTH). При характеристике новых препаратов фактора IX необходимо выполнять методики для определения активности в зависимости от анализируемого образца в сравнении со стандартным образцом фактора IX Всемирной организации здравоохранения. В случае, когда наблюдаются значительные расхождения показателей активности (то есть вариабельность анализируемых показателей связана с особенностями аналитической методики), необходимо доказать, что методика, выбранная для определения активности, обеспечивает сопоставимость с соответствующим ранее зарегистрированным не модифицированным препаратом путем сравнения уровней функциональной активности препаратов в тестах in vitro и in vivo. В плане управления рисками должны быть отражены вопросы, касающиеся последующего мониторинга лабораторных показателей уровня препарата в плазме. Соответствующие сведения должны быть доведены до потребителей данного препарата.
6.3. Оценка эффективности при исследовании ранее получивших лечение пациентов в возрасте 12 лет и старше
Фармакокинетические исследования
45. Фармакокинетические исследования должны проводиться, по крайней мере, с участием 12 ранее получавших лечение пациентов (получивших более 150 дневных введений препарата), страдающих гемофилией В (активность фактора IX < 2% от референтных значений), которые являются иммунокомпетентными (то есть без проявлений иммунодефицита) (у ВИЧ-инфицированных пациентов содержание CD4+ лимфоцитов должно составлять более 200 клеток/мкл). При исследовании должны оцениваться следующие показатели:
возрастающий уровень восстановления активности фактора;
период полувыведения in vivo;
площадь под кривой (AUC) и клиренс.
46. У исследуемых пациентов не должно наблюдаться спонтанных кровотечений и должны отсутствовать ингибиторы. Пациенты не должны быть младше 12 лет и не должны получать инфузионно любой препарат фактора IX в течение, как минимум 4 дней. Для того чтобы оценить индивидуальный ответ пациента, до первого введения нового препарата фактора IX, необходимо проанализировать информацию о фармакокинетике предыдущего препарата фактора IX (данные «исторического контроля») или последние данные о восстановлении активности и периоде полувыведения препарата). Образцы крови необходимо брать непосредственно перед введением препарата фактора IX в дозе 50 - 75 МЕ/кг (исходный уровень), через 10-15 минут (время относится к интервалу после завершения инфузии), через 30 минут и 1 час. Дополнительные сроки забора образцов включают 3, 6, 9, 24, 48 и 50 часов после инфузии. Взятие образцов через 72 часа не является обязательным, если пациенту было введена доза не менее 75 МЕ/кг. В зависимости от вида препарата фактора IX (например, препарат с удлиненным периодом полувыведения) точки забора образцов могут быть скорректированы с целью адекватной оценки временного профиля восстановления активности. В исследовании должно быть использовано не менее 3 серий препарата. Необходимо определять показатель восстановления активности фактора зарегистрированное в первый час после инфузии (выражается в [МЕ/мл]) или нарастающее восстановление активности фактора, то есть максимальное содержание фактора, зарегистрированное в первый час после инфузии (выраженное как [МЕ/мл]/[МЕ/кг]).
47. В материалах по результатам клинических исследований должен быть описан метод, используемый для анализа. Оптимально использовать один и тот же метод для оценки содержания фактора IX в лекарственном препарате и образцах плазмы крови пациента. Важно учитывать точный временной интервал после инфузии, в который фактически проводили отбор образцов, и использовать указанные точные значения при анализе результатов.
48. В отчет о клиническом исследовании необходимо включить результаты дополнительного анализа фармакокинетических исследований с учетом массы тела пациентов (нормальный диапазон, избыточная или недостаточная масса тела).
49. Пациенты, принимающие участие в фармакокинетическом исследовании, должны продолжать лечение препаратом после первого исследования. Через 3 - 6 месяцев применения препарата в тех же дозах, что и в первом исследовании, у них должны быть повторно определены те же фармакокинетические параметры. Кроме того, необходимо тестирование образцов на наличие ингибиторов (в соответствии с дизайном исследования, описанном в приложении № 3 к настоящей главе).
50. При проведении исследований необходимо учитывать, что препараты фактора IX выпускаются в разной дозировке, поэтому после восстановления концентрация активного вещества в растворе будет значительно различаться. В связи с этим необходимо исследовать фармакокинетику препарата фактора IX с самой низкой и самой высокой концентрацией, если иное не обосновано.
Эффективность препаратов фактора IX, включая эффективность при хирургическом вмешательстве
51. Оценка клинической эффективности препарата фактора IX должна проводиться как минимум у 20 ранее получивших лечение пациентов в возрасте 12 лет и старше, страдающих гемофилией В (активность фактора IX < 2%) и получавших введение препарата фактора IX более 150 дней, которые являются иммунокомпетентными (у ВИЧ-инфицированных пациентов содержание CD4+ лимфоцитов должно составлять более 200 клеток/мкл). В течение периода наблюдения необходимо оценить клинический ответ пациентов на воздействие не менее 50 введений препарата. Врач оценивает ответ, определяемый как «отсутствует», «умеренный», «хороший» или «отличный», у тех пациентов, которые получали препарат, находясь на лечении в стационаре по поводу купирования обильных кровотечений. Кроме того, врач должен определить ответ как минимум у 5 пациентов, у которых было по меньшей мере 10 хирургических вмешательств (включая обширные операции), с оценкой эффективности гемостаза, потери крови и потребностей в переливаниях крови. Для оценки клинической эффективности препарата фактора IX в отношении долгосрочной профилактики, пациентов следует лечить в течение 6 месяцев и регистрировать частоту и интервалы между кровоизлияниями, количество курсов лечения.
52. Клиническая эффективность оценивается путем расчета потребления фактора IX, выраженного как количество инфузий и величины МЕ/кг в месяц и в год, а также МЕ/кг на один случай применения препарата (профилактика спонтанных кровотечений; применение по требованию, то есть введение препарата для купирования уже развившегося кровотечения или хирургическое вмешательство).
Непрерывная инфузионная терапия
53. В случае если требуется непрерывная инфузионная терапия, исследование должно проводиться, как минимум у 10 пациентов с тяжелой формой гемофилии В (активность фактор IX < 2%), которым в плановом порядке проводятся обширные хирургические операции.
54. Перед операцией каждому пациенту, для того чтобы определить значение клиренса, необходимо провести фармакокинетические исследования. По величине клиренса рассчитывается начальная скорость инфузии лекарственного препарата по следующей формуле (при необходимости с допуском на соответствующую границу безопасности):
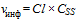 ,
,
где:
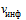 - скорость инфузии (МЕ/кг х ч);
- скорость инфузии (МЕ/кг х ч);
 - клиренс (мл/кг х ч);
- клиренс (мл/кг х ч);
 - желаемая равновесная концентрация (МЕ/мл).
- желаемая равновесная концентрация (МЕ/мл).
55. После первых 24 часов непрерывной инфузии необходимо ежедневно повторно рассчитывать клиренс, используя уравнение равновесного состояния, измеренное содержание и известную скорость инфузии.
56. В отчет о клиническом исследовании должны быть включены материалы, отражающие результаты оценки безопасности и эффективности препарата фактора IX во время операции и в течение, как минимум 6 дней после операции. Кроме того, должны быть приведены сведения о параметрах фармакокинетики с описанием используемого метода анализа, информация о суточной дозе фактора IX с указанием пути и скорости введения, сведения о потреблении фактора, гемостатическом ответе и кровопотере, о потребности в переливании крови, а также данные с описанием местных и системных нежелательных реакций.
57. В модуль 3 регистрационного досье должны быть включены фармацевтические данные по восстановлению и стабильности препарата фактора IX.
6.4. Отдельные вопросы клинических исследований с участием детей в возрасте 12 лет и старше, ранее получавших лечение
Выбор пациентов
58. В исследования включают ранее получавших лечение пациентов, получивших как минимум в течение 150 дней введение любого из препаратов фактора IX, которые рассматриваются, как пациенты с низким уровнем риска в отношении проявлений иммуногенности препарата.
59. Указанные ранее получившие лечение пациенты должны быть в возрасте 12 лет и старше, с уровнем содержания фактора IX < 2% и без признаков иммунодефицита (у ВИЧ-положительных пациентов содержание CD4-лимфоцитов должно составлять не менее 200 клеток/мкл). Вирусный статус пациентов должен быть охарактеризован и подтвержден документально. В исследование включают ВИЧ-отрицательных пациентов или пациентов с вирусной нагрузкой менее 200 частиц/мкл или менее 400000 копий/мл. В связи с более низкой частотой встречаемости гемофилии В по сравнению с гемофилией А в исследование должно быть включено как минимум 20 пациентов, регулярно получающих лечение препаратом фактора IX (как минимум 50 дней введения), и что должно быть подтверждено документально. Эти данные должны быть включены в регистрационное досье.
Оценка иммуногенности
60. Определение титра ингибиторов фактора IX проводится в соответствии с графиком, приведенным в приложении № 3 к настоящей главе. В процессе исследования взятие образцов для определения ингибиторов следует проводить не ранее, чем через 3 дня после введения препарата (если это возможно). Для исключения искажающего влияния на результаты определения ингибиторов остаточного содержания в исследуемых образцах плазмы препарата фактора IX, необходимо учитывать специфические свойства препарата, например, удлиненный период полувыведения. В отчет о клиническом исследовании должна быть включена полная информация о всех пациентах, у которых выявлены ингибиторы, содержащая сведения о клинической значимости, частоте выявления и количестве дней введения препарата. Для определения ингибиторов может быть использован метод Бетесда или метод Бетесда в модификации Неймегена, титр ингибиторов указывают в единицах Бетесда (БЕ). Образцы плазмы крови пациентов, в которых выявлены ингибиторы или имеется подозрение на наличие ингибиторов, должны храниться до конца клинического исследования и его оценки уполномоченным органом (экспертной организацией) государства-члена, что обеспечивает возможность повторного определения ингибиторов в случае необходимости.
Вирусная безопасность
61. Для всех лекарственных препаратов, получаемых из плазмы крови человека, необходимо соблюдение указаний в отношении вирусной безопасности, изложенных в разделе 5.2 настоящей главы. Подтверждающие материалы должны быть представлены в модуле 3 регистрационного досье лекарственного препарата.
62. Образцы сыворотки всех включенных в клиническое исследование пациентов, полученные до лечения, должны храниться при температуре минус 70°С для того, чтобы при необходимости провести их повторное тестирование.
6.5. Клинические исследования с участием детей младше 12 лет
63. Учитывая отличия реакций на введение препарата у детей и взрослых, необходимо проведение многоцентрового клинического исследования с участием детей. Поскольку встречаемость заболевания гемофилией В более низкая по сравнению с гемофилией А, количество включаемых в исследование детей должно составлять не менее 20, распределенных на 2 возрастные когорты. Как минимум 10 детей должны быть в возрасте от 6 до 12 лет, и как минимум 10 детей - младше 6 лет, которые ранее получали лечение препаратами фактора IX (более 50 дней введения). Клинические исследования, с участием детей младше 12 лет, проводятся только после того, как будет доказана безопасность применения препарата (в течение 50 дней введения) у 10 пациентов в возрасте от 12 лет и старше, которые были включены в исследование, как ранее получавшие лечение пациенты.
64. Клинические исследования с участием детей проводятся поэтапно и начинаются с оценки фармакокинетики (восстановление активности, период полувыведения in vivo, AUC и клиренс) у 10 пациентов каждой возрастной когорты. Для того чтобы адекватно оценить индивидуальный ответ пациента, до введения нового исследуемого препарата фактора IX, должна быть доступна информация о фармакокинетическом профиле ранее вводимого препарата фактора IX («исторические данные» или результаты недавно проведенного исследования с учетом показателей восстановления и периода полувыведения). Для удобства пациентов количество взятия образцов может быть уменьшено, временные точки для оценки фармакокинетики могут быть следующими: непосредственно перед введением препарата (исходный уровень), через 1 час, 10 часов, 24 часа и 48 часов после введения препарата. В зависимости от свойств препарата фактора IX (например, препарат с удлиненным периодом полувыведения), требуются дополнительные временные точки взятия образцов. В процессе проведения исследований возможны некоторые отклонения от данных указаний, поэтому необходимо зафиксировать точное время фактического взятия образцов после введения препарата и учитывать его при анализе результатов исследования. Оптимальным является проведение анализов образцов крови в центральной лаборатории, что снижает вариабельность результатов исследований.
65. У всех детей, участвующих в исследовании, необходимо контролировать потребление фактора IX (доза/кг для профилактики и терапии (по требованию, например для купирования кровотечения)), а также выработку ингибиторов. Определение ингибиторов должно проводиться в соответствии графиком, представленным в приложении № 3 к настоящей главе. При подозрении на возможность выработки ингибиторов исследования проводятся в соответствии с положениями, изложенными в разделе 5.3 настоящей главы. Для предрегистрационных клинических исследований детей в возрасте 12 лет и старше, ранее получавших лечение, исследование должно продолжаться до тех пор, пока пациенты не получат исследуемый лекарственный препарат как минимум в течение 50 дней введения.
В отчет о клиническом исследовании должна быть включена полная информация о всех пациентах, у которых выявлены ингибиторы, с оценкой их клинической значимости, указанием частоты выявления и количества дней введения препарата.
66. Для определения ингибиторов используется метод Бетесда или метод Бетесда в модификации Неймегена, титр ингибиторов указывается в единицах Бетесда (БЕ). Образцы плазмы крови пациентов, у которых выявлены ингибиторы или имеется подозрение на наличие ингибиторов, должны храниться, чтобы в случае необходимости можно было провести дополнительные исследования.
67. В регистрационное досье должны быть включены результаты оценки фармакокинетики (показатели нарастающего восстановления, периода полувыведения in vivo, AUC и клиренса), а также окончательные результаты оценки безопасности и эффективности у 20 детей (в возрасте от 0 до 12 лет), получивших исследуемый препарат в течение 50 дней введения.
68. В пострегистрационные исследования включают пациентов независимо от их возраста, ранее получавших лечение (более 150 дней введения), при условии, что клиническое исследование с участием детей в возрасте младше 12 лет было завершено до регистрации препарата.
6.6. Клинические исследования с участием ранее не получавших лечение пациентов
69. Ранее не получавшими лечение пациентами являются пациенты, которые никогда не получали лечение препаратами свертывания крови (за исключением предыдущего применения компонентов крови). Проведение клинических исследований с участием ранее не получавших лечение пациентов требуется в зависимости от типа изучаемого препарата фактора IX (например, новый препарат на основе модифицированного белка, характеризующийся удлиненным периодом полувыведения). В отношении препаратов фактора IX, полученных из плазмы, при использовании новых способов производства необходимость проведения исследований с участием ранее не получавшим лечение пациентов определяется в индивидуальном порядке. Для новых препаратов, требующих исследования у ранее не получавших лечения пациентов, информацию об отсутствии таких данных необходимо отразить в разделе 4.2 общей характеристики лекарственного препарата. Дозировка и способ введения (при включении информации в инструкцию по медицинскому применению) не утверждаются уполномоченным органом (экспертной организацией) государства-члена до тех пор, пока не будут получены данные об эффективности и безопасности по 20 ранее не получавшим лечение пациентам. Показания для ранее не получавших лечение пациентов основываются на результатах предрегистрационных клинических исследований с оценкой эффективности и безопасности, как минимум у 20 ранее не получавшим лечение пациентам, получивших препарат не менее чем в течение 50 дней введения, и при условии обязательного последующего наблюдения в пострегистрационном периоде, по меньшей мере 20 - 40 ранее не получавшим лечение пациентам, получавших препарат не менее 100 дней введения (из указанного числа пациентов, 20 - участвовавшие в исследовании по оценке эффективности и безопасности и 20 - новые пациенты).
70. Клинические исследования детской популяции с участием ранее не получавших лечение пациентов необходимо начинать после того, как будут завершены и проанализированы результаты исследований 10 пациентов в возрасте до 12 лет, из которых как минимум 5 пациентов, должны быть в возрасте до 6 лет, получивших по 50 дней введения препарата. Также должны быть завершены фармакокинетические исследования с участием детей младше 12 лет.
6.7. Пострегистрационные исследования
71. Для сбора дополнительных клинических данных и обеспечения согласованности в долгосрочной перспективе результатов предрегистрационных клинических исследований и рутинного применения должны быть проведены пострегистрационные исследования препарата фактора IX.
72. Протокол клинических исследований должен быть представлен в рамках плана управления рисками и включен в материалы регистрационного досье, в соответствии с требованиями Правил практики фармаконадзора.
73. Результаты исследований, проведенных до регистрации, должны быть учтены при разработке программы пострегистрационного исследования. Помимо таких показателей, как общая безопасность лекарственного препарата и его клиническая эффективность, особое внимание необходимо уделить таким вопросам иммуногенности как формирование ингибиторов, анафилактические реакции и тромботические осложнения.
74. Необходимо включить в исследование добровольцев тех регионов, где предполагается применять препарат фактора IX. Условием для включения пациентов в исследование является наличие документальных данных (истории болезни или амбулаторной карты, дневника, журнала), содержащих информацию за последние 50 дней приема лекарственного препарата или предыдущих 2 года. Это позволит охарактеризовать способ лечения конкретного пациента (профилактика, применение препарата по требованию для купирования кровоизлияний или при недавнем хирургическом вмешательстве). Пациенты с тяжелой гемофилией после успешной терапии, проведенной для индукции иммунологической толерантности (ИИТ), могут быть включены в исследование с целью получения информации об этой группе пациентов. Доля пациентов с индукцией иммунологической толерантности не должна превышать 25% всей когорты.
75. В пострегистрационное исследование препарата фактора IX для оценки его иммуногенного потенциала (помимо дополнительной оценки общей эффективности и безопасности), как правило, требуется включение 50 пациентов.
76. Включение в клиническое исследование препаратов фактора IX, полученных из плазмы (например, изготовленных по известной технологии), меньшего количества пациентов, необходимо обосновать. В исследование должны быть включены ранее получавшие лечение пациенты, получившие более 150 дней введения препарата, независимо от их возраста, следует обеспечить максимально сбалансированное распределение пациентов по возрасту. Допускается чтобы все пациенты, участвующие в предрегистрационных клинических исследованиях включались в последующие пострегистрационные исследования.
77. Протокол пострегистрационного исследования должен быть включен в регистрационное досье как часть плана управления рисками и одобрен уполномоченным органом (экспертной организацией) государства-члена в ходе регистрации препарата. Промежуточный отчет о выполнении исследований должен быть представлен уполномоченному органу (экспертной организации) государства-члена не позднее чем через 2 года после регистрации препарата, что дает возможность оценить скорость и правильность выбора пациентов, ход выполнения, результативность и соблюдение сроков проведения исследования. Пострегистрационное исследование должно быть завершено в течение 4 лет после регистрации препарата фактора IX.
78. Требования, предъявляемые к дизайну пострегистрационного исследования, приведены в приложении № 3 к настоящей главе.
6.8. Клинические исследования при изменениях процесса производства
79. Изменения, внесенные в процесс производства, могут привести к значительным изменениям свойств препарата фактора IX, таким как изменение структуры и активности фактора свертывания. Необходимо изучить влияние внесенных изменений в производственный процесс (например, изменения стадий инактивации вирусов или способов очистки) на биологические свойства и активность препарата. В случае если нельзя исключить значительное влияние внесенных изменений на активность фактора свертывания, должны быть представлены данные по оценке фармакокинетики, безопасности и эффективности препарата. Эти данные должны быть получены путем проведения исследований сопоставимости показателей качества препаратов фактора IX, полученных до и после внесения изменений в процесс производства. Указанные исследования проводятся в соответствии с главами 9.1 и 9.2 настоящих Правил.
Общие аспекты клинических исследований
80. При внесении изменений в процесс производства препарата фактора IX держатель регистрационного удостоверения должен подтвердить, что препараты фактора IX, произведенные до и после внесения изменений в процесс производства, сопоставимы в отношении безопасности, эффективности и качества. Исследования по доказательству сопоставимости проводят поэтапно, начиная с исследований по оценке качества, которые при необходимости должны быть подтверждены результатами доклинических или и клинических исследований.
81. Объем данных клинических исследований, которые должны быть представлены, определяется в каждом конкретном случае в зависимости от потенциального влияния изменений, внесенных в процесс производства на свойства препарата фактора IX. Объем этих данных может варьировать от сравнительных фармакокинетических исследований препаратов фактора IX, полученных до и после внесения изменений в процесс производства, до полного объема клинических исследований, требуемых для нового препарата фактора IX (в соответствии с настоящим разделом).
82. В регистрационном досье должны быть представлены данные о сохранении профиля иммуногенности препарата фактора IX, полученного после внесения изменения в процесс производства, по сравнению с препаратом, изготовленным до внесения изменения. В зависимости от ожидаемого риска может потребоваться проведение клинических исследований с перекрестным дизайном для подтверждения сопоставимости препаратов, полученных до и после внесения изменения в процесс производства.
83. Материалы по результатам исследования сопоставимости препаратов фактора IX, полученных до и после внесения изменения в процесс производства, должны отражать оценку потенциального воздействия внесенных изменений на безопасность и эффективность данного препарата, что служит обоснованием программы клинической разработки препарата фактора IX.
Эффективность препаратов фактора IX
84. При изменениях в процессе производства должны быть представлены доказательства, подтверждающие, что внесенные изменения не повлияли на фармакокинетику препарата фактора IX. Указания по данному вопросу приведены в главе 9.2 настоящих Правил и правилах исследований биоэквивалентности лекарственных препаратов в рамках Союза.
85. Сравнительное фармакокинетическое исследование препаратов, полученных до и после внесения изменений в процесс производства, должно проводиться с участием, по меньшей мере 12 ранее получавших лечение пациентов, страдающих гемофилией В (фактор IX < 2%). При проведении исследования необходимо регистрировать такие показатели, как нарастающее восстановление, период полувыведения in vivo, площадь под кривой (AUC) и клиренс. У пациентов должны отсутствовать ингибиторы к фактору IX, и не должно быть спонтанных кровотечений. В исследование могут быть включены пациенты от 12 лет и старше, которым не вводили ни один из препаратов фактора IX в течение как минимум 4 дней (отмывочный период), для исключения его влияния на оцениваемые показатели. Отбор образцов крови следует осуществлять непосредственно перед инъекцией дозы 50 - 75 МЕ/кг препарата фактора IX (исходный уровень), через 10-15 минут (временные точки обозначают временной интервал после завершения инфузии), через 30 минут и 1 час. Дополнительными временными точками являются 3, 6, 9, 24, 48 и 50 часов после инфузии. Отбор образцов через 72 часа является дополнительным при условии, что пациент получил не менее 75 МЕ/кг.
86. В зависимости от вида препарата фактора IX (например, с удлиненным периодом полувыведения) могут потребоваться дополнительные временные точки отбора образцов. В клиническом исследовании следует использовать как минимум 3 разные серии препарата, полученные после внесения изменений в процесс производства. Нарастающее восстановление определяется как максимальное содержание фактора, зарегистрированное через 30 минут после инфузии, представленное в МЕ/мл или МЕ/кг.
87. Необходимо регистрировать точное время после инфузии, в которое фактически проводился отбор образцов, и использовать указанные точные значения при анализе результатов конкретных образцов.
88. Пациенты, принимающие участие в фармакокинетическом исследовании, должны продолжать лечение препаратом фактора IX, полученным после внесения изменений в процесс производства, в течение 6 месяцев. Через 3-6 месяцев лечения препаратом фактора IX в той же дозе, что и в первом исследовании у пациентов должны быть повторно определены те же фармакокинетические параметры.
89. Если кому-либо из пациентов, участвующих в клинических исследованиях, потребуется хирургическое вмешательство, ответ на лечение препаратом фактора IX должен оцениваться врачом, включая эффективность гемостаза, кровопотерю, потребность в переливаниях крови и развитие тромбоэмболических осложнений.
6.9. План управления рисками
90. План управления рисками для конкретного препарата должен быть составлен с учетом результатов предрегистрационных исследований, и с учетом общих указаний по формированию плана управления рисками. В настоящем разделе указаны показатели, которые должны быть отражены в плане управления рисками, но они не должны рассматриваться, как исчерпывающие. Далее приведен перечень показателей, касающихся информации о новых препаратах фактора IX, а также препаратах фактора IX, в процесс производства которых внесены значительные изменения и которые должны быть проанализированы в соответствующих разделах плана управления рисками.
91. План управления рисками разрабатывается в соответствии с положениями Правил практики фармаконадзора.
92. Протокол пострегистрационного исследования необходимо включить в соответствующее дополнение плана управления рисками.
Формирование ингибиторов фактора IX
93. Наиболее серьезным осложнением при гемофилии является образование ингибиторов у ранее не получавших и ранее получавших лечение пациентов, однако формирование ингибиторов при гемофилии В наблюдается реже, чем при гемофилии А. Тщательно проведенный анализ зарегистрированных de novo ингибиторов и ингибиторов транзиторных (определяемых периодически), должен быть представлен в части VII плана управления рисками в виде сводного отчета. В отчет должна быть включена следующая информация о:
источнике сообщения об ингибиторах (например, отчеты клинических исследований, пострегистрационный мониторинг, спонтанные сообщения);
низких или высоких титрах периодически выявляемого ингибиторе (прежде чем сделать окончательное заключение о наличии ингибиторов, каждый положительный лабораторный тест должен быть подтвержден путем повторного анализа в центральной лаборатории с использованием второго отдельно взятого образца от одного и того же пациента. Образцы необходимо хранить с целью возможности их последующего тестирования);
ингибиторах фактора IX 1-го или 2-го типов.
94. Факторы риска образования ингибиторов фактора IX:
тяжесть гемофилии;
терапевтический статус (ранее не получавший лечение пациент или ранее получавший лечение пациент);
кумулятивное воздействие препаратов фактора IX (общее количество дней введения препарата и доза на одно введение);
вид мутации гена;
этническая принадлежность пациента;
возраст пациента при начале терапии;
интенсивность терапии;
частота формирования ингибиторов с определением 95% доверительного интервала (ДИ);
отдельные группы пациентов;
пациенты, перенесшие хирургическое вмешательство, у которых впоследствии сформировались ингибиторы;
любой конкретный риск (например, формирование ингибиторов, отсутствие клинического эффекта), связанный с заменой одного препарата на другой препарат фактора IX, должен быть проанализирован отдельно. Анализ риска критически важен для препаратов фактора IX в случае внесения значительных изменений в процесс их производства. Замена у пациента препарата, произведенного до внесения изменений, на препарат, изготовленный после внесения изменений в процесс производства, требует тщательного изучения.
Отсутствие клинического эффекта на применение фактора IX
95. Отсутствие клинического эффекта на применение фактора IX от лечения препаратом фактора IX и развитие кровотечения могут указывать на формирование ингибиторов. Важное значение имеет учет развития ожидаемых нежелательных реакций. Необходимо тщательное наблюдение за пациентами, включая оценку ингибиторов (потребление, восстановление, период полувыведения, тестирование на ингибиторы).
Реакции гиперчувствительности, анафилактические реакции
96. При применении препаратов фактора IX возможно развитие реакций гиперчувствительности и анафилактических реакций, в том числе, реакций на белки клеток-хозяина, вспомогательные вещества или реагенты, используемые в процессе производства таких препаратов. Указанные реакции необходимо классифицировать в соответствии с местными и системными реакциями гиперчувствительности. Пациентов, у которых развилась анафилактическая реакция, необходимо тщательно обследовать и контролировать на выработку ингибиторов. Должна быть заполнена соответствующая анкета или другая форма отчетности, в которой необходимо указать информацию о статусе терапии (например, ранее не получавшие лечение пациенты или ранее получавшие лечение пациенты). Должны быть представлены данные о характеристике класса иммуноглобулинов антител к фактору IX, определяемых с использованием соответствующих методов (например, антитела класса IgE, IgG).
Тромбогенность
97. Тромботические осложнения необходимо отслеживать и сообщать о них.
98. Поскольку на определение уровня содержания (активности) фактора IX в плазме существенно влияет метод, используемый при клиническом мониторинге, в тех случаях, когда наблюдается расхождение результатов анализа между данными клинического исследования и данными посрегистрационного мониторинга (что зависит от методики, используемой при пострегистрационном мониторинге (в соответствии с подразделом 6.2 настоящего раздела)), в сведения о препарате фактора IX должна быть включена данная информация. Однако допускаются также и другие подходы, включая использование обучающих материалов для подготовки клинических лабораторий. В плане управления рисками должны быть приведены сведения, обеспечивающие устранение риска несоответствия результатов мониторинга при определении уровня фактора IX в плазме, и информация о мерах, направленных на предотвращение такого несоответствия.
ПРИЛОЖЕНИЕ № 1
к главе 26 Правил проведения
исследований биологических
лекарственных средств
Евразийского экономического союза
ТРЕБОВАНИЯ
к дизайну клинических исследований

ПРИЛОЖЕНИЕ № 2
к главе 26 Правил проведения
исследований биологических
лекарственных средств
Евразийского экономического союза
ТРЕБОВАНИЯ
к выбору изучаемых параметров в клинических исследованиях новых препаратов фактора IX
| Участники исследования | Вид клинического исследования | Изучаемые параметры |
|---|---|---|
| Предрегистрационные исследования у ранее получавших лечение пациентов в возрасте 12 лет и старше | ||
12 пациентов с гемофилией В (ранее получавшие лечение пациенты в возрасте 12 лет и старше, фактор IX  2%) без ингибиторов и без спонтанных кровотечений)
2%) без ингибиторов и без спонтанных кровотечений)
|
фармакокинетика1 | показатель нарастающего восстановления, период полувыведения, AUC, клиренс. Пациенты должны пройти повторное тестирование через 3 - 6 месяцев (включая анализ на ингибиторы фактора IX) |
| безопасность | артериальное давление, частота сердечных сокращений, температура, частота дыхания и нежелательные реакции, тромбогенность | |
5 пациентов с гемофилией В (ранее получавшие лечение пациенты в возрасте 12 лет и старше,
(фактор IX  2%), перенесших, по меньшей мере, 10 хирургических вмешательств
2%), перенесших, по меньшей мере, 10 хирургических вмешательств
|
клиническая эффективность | эффективность гемостаза, кровопотеря и потребности в переливании крови, потребление фактора IX |
| безопасность | нежелательные реакции, тромбогенность | |
Эффективность и безопасность у 20 ранее получавших лечение пациентов (в возрасте 12 лет и старше, фактор IX  2% и CD4 более 200 клеток/мкл)
2% и CD4 более 200 клеток/мкл)
|
клиническая эффективность | потребление фактора IX, оценка врачом ответа при лечении обильных кровотечений |
| иммуногенность | титр ингибиторов в единицах Бетесда непосредственно перед первым введением препарата, на 10 - 15 день введения, на 50 - 75 день введения и в случае каких-либо подозрений на образование ингибиторов. Продолжительность введения препарата фактора IX в течение, как минимум, 50 дней | |
| безопасность | нежелательные реакции, тромбогенность | |
| Предрегистрационные исследования у детей младше 12 лет2 | ||
10 пациентов с гемофилией В (ранее получавшие лечение пациенты от 6 до 12 лет, фактор IX  2%) без ингибиторов и без спонтанных кровотечений и 10 пациентов с гемофилией В (более 50 дней введения препарата, младше 6 лет, фактор IX
2%) без ингибиторов и без спонтанных кровотечений и 10 пациентов с гемофилией В (более 50 дней введения препарата, младше 6 лет, фактор IX  2%) без ингибиторов и без спонтанных кровотечений
2%) без ингибиторов и без спонтанных кровотечений
|
фармакокинетика | показатель нарастающего восстановления, период полувыведения, AUC, клиренс |
| безопасность | артериальное давление, частота сердечных сокращений, температура, частота дыхания и нежелательные реакции, тромбогенность | |
| Многоцентровое исследование с участием 20 детей с гемофилией В, разделенных на 2 возрастные подгруппы, включающие 10 ранее получавших лечение пациентов (от 6 до 12 лет) и 10 детей (младше 6 лет, получивших более 50 дней введения препарата) | клиническая эффективность | потребление фактора IX, оценка врачом ответа при лечении обильных кровотечений |
| иммуногенность | тестирование на наличие ингибиторов непосредственно перед первым введением препарата, на 10 - 15 день введения, на 50 - 75 день введения и в случае каких-либо подозрений на образование ингибиторов. Продолжить введение препарата необходимо в течение, как минимум, 50 дней | |
| безопасность | нежелательные реакции, тромбогенность | |
| Пострегистрационные исследования | ||
| 50 ранее получивших лечение пациентов получают в общей сложности 100 дней введения препарата3 и 50 ранее не получивших лечение пациентов получают в общей сложности 100 дней введения препарата4 | клиническая эффективность, иммуногенность и безопасность | необходимо представить протокол в соответствии с указаниями, приведенными в приложении № 3 к настоящей главе |
| 20 ранее не получавших лечение пациентов в возрасте до 12 лет с проведением терапии в течение по меньшей мере 50 дней введения препарата фактора IX5 | клиническая эффективность | потребление фактора IX, оценка врачом ответа на лечение обильных кровотечений |
| иммуногенность | тестирование на наличие ингибиторов непосредственно перед первым введением препарата, на 10 - 15 день введения, на 50 день введения и в случае каких-либо подозрений на образование ингибиторов. Продолжить введение препарата необходимо в течение, как минимум, 50 дней | |
| безопасность | нежелательные реакции, артериальное давление, частота сердечных сокращений, температура, тромбогенность | |
------------------------------
1 Для оценки индивидуального ответа пациента, до первого введения нового препарата фактора IX должна быть доступна информация по фармакокинетике, например, данные оценки фармакокинетических параметров у пациента при предыдущем применении препарата фактора IX (по крайней мере, данные по восстановлению активности и периоду полувыведения - исторические или недавно полученные).
2 Исследования начинают после получения и анализа результатов исследования у 10 ранее получавших лечение пациентов в возрасте 12 лет и старше, получивших 50 дней введения препарата фактора IX.
3 Ранее получавшие лечение пациенты из предрегистрационного исследования могут продолжать участвовать в исследовании до 100-го дня введения препарата. Новые ранее получавшие лечение пациенты должны получить не менее 100 дней введения препарата.
4 По меньшей мере 20 - 40 ранее не получавших лечение пациентов должны находиться под наблюдением до получения 100 дней введения препарата фактора IX (из этих пациентов не менее 20 ранее не участвовали в предрегистрационных исследованиях фактора IX).
5 Исследование с участием ранее не получавших лечение пациентов в возрасте до 12 лет может быть начато после завершения и анализа результатов предрегистрационного исследования у 10 пациентов в возрасте младше 12 лет, получавших лечение в течение 50 дней введения препарата фактора IX (не менее 5 из которых должны быть младше 6 лет), и после завершения исследований фармакокинетики у детей в возрасте от 0 до 12 лет.
------------------------------
ПРИЛОЖЕНИЕ № 3
к главе 26 Правил проведения
исследований биологических
лекарственных средств
Евразийского экономического союза
ДИЗАЙН
пострегистрационных исследований на формирование ингибиторов к фактору IX
Критерии включения пациентов в исследование
Критериями включения пациентов в исследование являются:
диагноз гемофилии В;
активность фактора IX  2% от референтных значений;
2% от референтных значений;
количество дней введения препарата фактора IX перед включением в исследование более 150;
иммунокомпетентный статус пациента (то есть без проявлений иммунодефицита, CD4-лимфоциты более 200 клеток/мкл, ВИЧ-отрицательный пациент или ВИЧ-положительный пациент, имеющий вирусную нагрузку менее 200 частиц/мкл или не более 400000 копий/мл.
Ранее получивших лечение пациентов каждой возрастной группы необходимо включать в исследование при условии, что исследования с участием детей (фармакокинетики, эффективности и безопасности) завершены, отчет об исследовании представлен и одобрен уполномоченным органом (экспертной организацией) государства-члена.
Документальное подтверждение характеристик пациента
В характеристику пациента включается следующая информация:
вид дефекта гена;
этническая принадлежность пациента;
семейный анамнез гемофилии;
вся информация об ингибиторах, образующихся в организме пациента;
вирусный статус пациента (пациент должны быть ВИЧ-отрицательным или иметь вирусную нагрузку менее 200 частиц/мкл или не более 400 000 копий/мл);
сведения о сопутствующих заболеваниях или сопутствующей фармакотерапии, которые могут существенно повлиять на систему свертывания крови или иммунореактивность (должна быть отражена любая информация, касающаяся данного вопроса).
Включение пациентов в исследование
В исследование включаются пациенты при соблюдении следующих условий:
включение в исследование как минимум, 50 пациентов для пострегистрационного исследования;
последующее наблюдение за каждым пациентом должно продолжаться до получения не менее 100 дней введения препарата фактора IX;
представление информации о ходе набора пациентов на регулярной основе (порядок предоставления информации должен быть согласован с уполномоченным органом (экспертной организацией) до утверждения процедуры);
представление промежуточного отчета о выполнении исследования уполномоченному органу (экспертной организации) государства-члена спустя 2 года после регистрации препарата, для оценки скорости и правильности подбора пациентов, хода выполнения, результативности и соблюдения сроков проведения исследования;
пострегистрационное исследование должно быть завершено в течение 4 лет после регистрации препарата фактора IX.
Порядок проведения исследования
Пациенты, включенные в исследование, не должны иметь клинических проявлений, указывающих на наличие ингибиторов, а восстановление и тест на ингибиторы должны быть подтверждены в центральной лаборатории, свидетельствуя о том, что у пациента при включении в исследование ингибиторы отсутствуют. В случае если тест на ингибиторы не отрицательный, в центральной лаборатории должно быть проведено повторное подтверждающее тестирование второго отдельно взятого образца. Схема исследований представлена в таблице.
Таблица
Схема исследований
| Оцениваемый параметр | Ранее используемый препарат1 | Исследуемый препарат ДВ2 1 | Исследуемый препарат ДВ 10 - 15 | Исследуемый препарат ДВ 50 - 75 | Исследуемый препарат ДВ ~ 100 |
|---|---|---|---|---|---|
| Ингибиторы3 | x | x4 | x | x | x |
| Восстановление активности фактора IX | x | x | x | x | x |
------------------------------
1 В исследование включаются новые пациенты, то есть пациенты у которых ранее исследуемый препарат не являлся препаратом фактора IX в отношении которого проводится данное пострегистрационное исследование.
2 ДВ - дни введения препарата фактора IX.
3 После завершения периода отмывки от предшествующего препарата фактора IX необходимо хранить резервный образец крови.
4 Необходимо повторить определение исходного уровня ингибиторов перед первым введением исследуемого препарата фактора IX.
------------------------------
При подозрении на наличие у пациента ингибиторов, необходимо проводить их тестирование.
По данным дневников пациентов оценивается общее количество дней введения препарата фактора IX в год и средняя доза на килограмм массы пациента в год (потребление).
Предполагаемый режим лечения для каждого пациента при включении в исследование и обоснование каждого дня введения препарата фактора IX должны быть документированы.
В случае кровотечения: документированные сведения, заключение о тяжести и результатах лечения врачом - клиницистом и пациентом (потребление).
В случае хирургической операции должны быть собраны другие данные (протокол операции) (например, вид операции (плановая или экстренная), документация об осложнениях, способ введения препарата фактора IX; потребление).
Необходимо проводить мониторинг всех нежелательных реакций.
Особенности определения уровня ингибиторов
Определение ингибиторов должно проводиться, когда уровень фактора IX в плазме достигает своего надира перед замещением (необходимо представить документальное подтверждение результатов исследования последней инфузии).
Для учета ингибиторов необходимо использовать анкеты (опросные листы) или другие формы отчетов. В случае лечения пациентов по требованию (например, при необходимости купирования кровотечения) ингибитор может остаться не выявленным, если пациенты не получали лечение в течение более 2 недель. После прекращения терапии уровень содержания ингибитора может постепенно снижаться в соответствии с периодом полувыведения (Т1/2) иммуноглобулинов. В случае положительного результата теста на ингибиторы, необходимо также исследование фармакокинетических параметров (показатель восстановления) для подтверждения ингибиторной активности.
Сопутствующая фармакотерапия. В настоящее время все пациенты могут принимать участие в исследованиях при условии, что они являются иммунокомпетентными (количество CD4-лимфоцитов более 200 клеток/мкл, ВИЧ-отрицательные или имеют вирусную нагрузку менее 200 частиц/мкл около 400000 копий/мл). Пациенты с ВИЧ-инфекцией получают интенсивную фармакотерапия, влияние которой не известно. Например, не известно может ли влиять высокоактивная антиретровирусная терапия на формирование ингибиторов или эффективность лечения. Аналогичные проблемы можно ожидать у пациентов с гепатитом С, некоторые из них получают терапевтические препараты, у других - отмечается более низкий уровень тромбоцитов, снижение функции печени и изменение коагуляции. Указанные пациенты могут быть включены в исследования для получения дополнительных данных об эффективности препаратов в этой конкретной группе пациентов, при этом необходимо собрать как можно больше информации по пациентам с сопутствующей патологией.
Глава 27. Требования по удалению, снижению концентрации или замене тиомерсала в вакцинах
1. Общие положения
1. Тиомерсал - антимикробный консервант, входящий в состав ряда вакцин, вводимых человеку. В дополнение к антимикробной функции, тиомерсал может влиять на антигенность и стабильность вакцины. Тиомерсал и другие ртутьорганические соединения в инактивированных вакцинах применяются:
на стадии получения готового нерасфасованного продукта для предотвращения его контаминации;
на более ранних стадиях производства в качестве инактивирующего средства или с целью предотвращения контаминации.
2. Тиомерсал и другие ртутьорганические соединения не используются при производстве живых вакцин, поскольку могут привести к инактивации действующего вещества. Производители и уполномоченные органы (экспертные организации) государств-членов, допускают использование данных антимикробных соединений, поскольку методы, использующиеся для стерилизации вакцин (температура и (или) стерилизующая фильтрация), в том числе методы, приведенные в Фармакопее Союза и в фармакопеях государств-членов, не могут применяться к действующим веществам вакцин.
3. Ртутьорганические консерванты могут использоваться в ходе производства активной фармацевтической субстанции, нерасфасованного промежуточного продукта, готового нерасфасованного продукта и (или) готовой вакцины.
1.1. Активные фармацевтические субстанции
4. В некоторых вакцинах, в которых тиомерсал используется в процессе производства активных фармацевтических субстанций, ртутьорганические соединения обеспечивают микробиологическую чистоту антигена (например, процедуры, предусмотренные стадией сбора вируса гриппа после культивирования на куриных эмбрионах, предусматривают использование антибактериального агента на данном этапе получения продукта, поскольку эффективность тиомерсала для этих целей подтверждена). Тиомерсал также используется при инактивации антигена (например, его добавляют в качестве дополнительного агента при тепловой инактивации коклюшной бактерии).
1.2. Нерасфасованные промежуточные продукты
5. В силу своей лабильности, вакцины не подвергаются финишной стерилизации. Для некоторых нерасфасованных компонентов комбинированных вакцин и нерасфасованных продуктов, представляющих собой суспензию, стерилизующая фильтрация непосредственно перед смешиванием или розливом неприменима. Таким образом, использование антимикробного консерванта является дополнительной гарантией отсутствия и распространения бактериального загрязнения.
1.3. Готовый нерасфасованный продукт и готовая вакцина (серия вакцины)
6. В некоторых случаях один готовый нерасфасованный продукт используется для производства как в многодозной первичной упаковке (присутствие консерванта в которой обязательно в соответствии с требованиями Фармакопеи Союза, а при отсутствии в ней - в соответствии с требованиями фармакопей государств-членов, так и однодозной первичной упаковке готовой вакцины. Использование однодозных вакцин считается принципиально более безопасным подходом по сравнению с многодозными вакцинами. При использовании однодозных первичных упаковок (контейнеров) (далее - контейнеры) для вакцин, как правило, не существует оснований для использования тиомерсала или любого другого консерванта. Многодозные контейнеры имеют преимущество с точки зрения простоты применения и стоимости в случае необходимости проведения кампаний массовых вакцинаций в сложных условиях (например, вакцинация больших групп населения в сжатые сроки). Несмотря на требование к наличию консерванта в вакцинах, разлитых в многодозные контейнеры, если после вскрытия упаковки ее содержимое будет использовано в течение ограниченного периода времени (например, в течение нескольких часов), особой необходимости в наличии консерванта нет. Однако безопасность использования многодозных контейнеров зависит не только от времени после вскрытия упаковки, но еще и от стерильности системы укупорки флакона, иглы, шприца и их надлежащего использования.
7. Фактором, позволяющим допустить применение консерванта, является мутность отдельных вакцин, которая может препятствовать визуальному выявлению в них микробной контаминации. Присутствие консерванта в таких вакцинах является гарантией отсутствия бактериального роста.
8. Для вакцинации младенцев и детей младшего возраста следует использовать вакцины без тиомерсала.
9. Снижение нагрузки вакцин ртутьорганическими консервантами достигается тремя основными путями:
снижением количества тиомерсала в готовой вакцине;
исключением тиомерсала из состава вакцины;
замены тиомерсала альтернативным консервантом.
Эти три варианта не являются взаимоисключающими, поскольку на начальном этапе производитель может заявить об изменении, связанном с уменьшением количества тиомерсала в вакцине, и при этом параллельно разрабатывать вакцину с заменой консерванта или вакцину, полностью свободную от консервантов, причем последний вариант является предпочтительным.
10. Вакцины, не содержащие ртутьорганических консервантов, могут быть получены путем исключения этих компонентов из всех стадий производства, предусматривающих их добавление. Вакцины со сниженным содержанием ртутьорганических соединений могут быть получены путем удаления данных соединений физико-химическими способами или путем исключения этих соединений на заключительном этапе производства. При этом пониженное содержание ртутьорганических веществ не будет выполнять функции антимикробного консерванта.
11. Снижение или удаление тиомерсала может оказывать влияние на микробиологическую чистоту, растворимость, антигенность, иммуногенность, реактогенность и стабильность вакцины. Таким образом, внесению данных изменений в технологию должны предшествовать серьезные исследования и валидационные мероприятия. Каждая вакцина должна рассматриваться в индивидуальном порядке. Возможное влияние на безопасность, эффективность и качество вакцины, должно оцениваться в каждом конкретном случае. После критического анализа, в некоторых случаях, может потребоваться проведение клинических исследований для изучения влияния вносимых изменений на безопасность и эффективность. Таким образом, весь процесс изменений следует рассматривать как средне- и долгосрочное мероприятие. В настоящей главе рассматриваются вопросы оценки безопасности, эффективности и качества вакцин, возникающие в связи с такими изменениями, а также объем представляемых для оценки документов и данных.
2. Снижение концентрации и (или) удаление тиомерсала из вакцин
12. Конечной целью производственного процесса должно являться получение вакцин без ртутьорганических консервантов. Однако сократить концентрацию консерванта в готовой вакцине до остаточных количеств можно в более краткосрочной перспективе. Это достижимо путем использования физико-химических методов удаления консерванта на промежуточных этапах производства или путем удаления или уменьшения его количества на стадии сведения компонентов при получении готового нерасфасованного продукта.
13. Процесс удаления ртутьорганических консервантов физикохимическими методами (аналогично требованиям к процессу удаления других химических веществ, используемых в процессе производства) должен быть описан, а эффективность используемых методов должна быть подтверждена не менее чем на трех сериях вакцины.
14. Величина остаточной концентрации консерванта должна быть установлена и указана в спецификации на вакцину. Если метод количественного определения консерванта не обладает достаточной чувствительностью (пределом количественного определения), то данная величина может быть установлена как расчетная. Наличие остаточного консерванта должно быть отражено в общей характеристике лекарственного препарата в соответствии с требованиями к инструкции по медицинскому применению лекарственного препарата и общей характеристике лекарственного препарата для медицинского применения.
3. Замена тиомерсала другими противомикробными средствами
15. Ртутьорганические консерванты могут быть заменены на альтернативные вещества, используемые в настоящее время при производстве вакцин. При этом альтернативное вещество должно обладать соответствующей антимикробной эффективностью. Представляемая документация должна включать результаты исследования по хранению не менее трех серий вакцин после замены консерванта, а также, при необходимости, результаты доклинических исследований.
16. Замену консерванта необходимо производить только после тщательного изучения соотношения «пользы - риска» в соответствии с требованиями к противомикробной эффективности, совместимости альтернативного вещества с антигеном (антигенами), вспомогательными веществами и первичной упаковкой, а также влиянием на стабильность, безопасность и эффективность вакцины.
17. Если ртутьорганические соединения будут заменены на вещество, используемое в качестве инактивирующего агента, исследование эффективности такой замены должно подтверждать, что инактивирующая способность нового соединения, по меньшей мере, эквивалентна эффективности ранее используемого соединения. Подобное исследование следует проводить по меньшей мере в трех независимых циклах инактивации.
18. Общие указания по включению противомикробных средств в вакцинах также содержатся в Фармакопее Союза, а при отсутствии в ней - в фармакопеях государств-членов.
4. Влияние снижения концентрации, удаления или замены тиомерсала на микробиологическую чистоту вакцины
19. Основной целью использования ртутьорганических консервантов или любых других антимикробных соединений - является обеспечение микробиологической чистоты вакцины (отсутствие бактериальной контаминации). Основной нежелательный эффект от исключения, замены консерванта или снижения его концентрации, может заключаться в ухудшении качества вакцины по таким показателям, как наличие посторонней микрофлоры, стерильность и содержание эндотоксинов. Валидация процесса снижения концентрации тиомерсала или его удаления (или, по возможности, замены) должна быть основана на данных по микробиологической чистоте продукта на соответствующих стадиях производства, включая все стадии, на которых антимикробное средство было исключено или заменено и других стадиях, где этот процесс может оказать влияние на качество вакцины.
20. Сведения о микрофлоре и уровню эндотоксинов должны содержать сравнительный анализ с данными, полученными ранее, до внесения в процесс производства изменений, связанных с консервантом. Такие сведения должны быть получены в результате испытаний, по меньшей мере, трех серий вакцины.
21. Данные о микробиологической чистоте после хранения должны включать в себя результаты исследования не менее трех серий вакцины.
5. Влияние снижения концентрации, удаления или замены тиомерсала на качество вакцины
22. Снижение концентрации тиомерсала, его удаление или замена на иной антимикробный консервант могут оказывать влияние не только на безопасность, но и на эффективность вакцины. Например, следы ионов ртутьорганических соединений, оказывают стабилизирующее воздействие на поверхностный антиген вируса гепатита В (HbsAg), который является действующим веществом рекомбинантной вакцины против гепатита. Также стабилизирующее влияние ртутьорганические соединения оказывают на все вакцины, содержащие клеточный коклюшный компонент. В целом, ртутьорганические консерванты могут взаимодействовать с антигенами в вакцине, и их снижение, удаление или замещение на некоторых или всех этапах производства может повлиять на качество антигена. Поэтому все изменения, касающиеся снижения, замены или удаления ртутьорганических соединений, должны подтверждаться соответствующими данными о стабильности вакцины и ее остальных характеристиках, включая данные об активности. В каждом конкретном случае следует рассмотреть возможность представления актуальных данных о стабильности, поскольку полный набор данных о стабильности может не понадобиться.
23. В случаях, когда подтверждения качества вакцины на соответствие спецификации недостаточно необходима более подробная характеристика структуры белка, профиля примесей и биологической активности. Все данные, характеризующие вакцину после внесения в ее состав изменений, должны быть получены в сравнении с составом вакцины, произведенной согласно утвержденной технологии. В тех случаях, когда тиомерсал исключен или заменен на заключительных стадиях производства, возможны сложности с изучением влияния данных изменений на антиген, поскольку концентрация антигена, вспомогательные вещества, а также адсорбция антигена на адъювантах, могут препятствовать или неспецифически влиять на результаты исследований. Также, на основе изученных характеристик, должна быть определена необходимость проведения соответствующих дополнительных доклинических и клинических исследований в сравнении с вакциной, содержащей тиомерсал. К некоторым вакцинам могут применяться положения главы 9.1 настоящих Правил.
6. Вопросы, касающиеся безопасности и эффективности вакцин
24. Несмотря на то, что удаление тиомерсала, добавляемого на стадии готовой вакцины (готовой партии), не должно в значительной степени влиять на ее эффективность, следует подтвердить, что вакцина имеет биологические характеристики, одинаково стабильные в сравнении с исходным составом. Необходимость проведения клинических исследований следует рассматривать в каждом конкретном случае.
6.1. Эффективность
25. Если клинические исследования сочтены необходимыми, в ходе их проведения требуется установить эквивалентность между вакциной содержащей тиомерсал и вакциной, не содержащей тиомерсал, используя соответствующие дизайны исследований. Заявитель должен продемонстрировать, что вакцина, не содержащая тиомерсала, не уступает исходной вакцине, содержащей тиомерсал. Терапевтическая эквивалентность между данными вакцинами должна быть установлена в соответствии с актами органов Союза в сфере обращения лекарственных средств.
26. Мощность исследований должна быть достаточной для испытания на эквивалентность. Если вакцина изначально разработана без содержания тиомерсала, нет необходимости устанавливать терапевтическую эквивалентность с антигенно схожей вакциной содержащей тиомерсал.
27. Доказательство иммуногенности вакцинного антигена вакцины, не содержащей тиомерсал, можно получить, если вакцинный антиген уже входит в состав такой комбинированной вакцины.
6.2. Безопасность
28. Удаление или снижение содержания тиомерсала может привести к негативному влиянию на безопасность вакцины и поэтому требует отдельного изучения. В ходе сравнительных исследований по изучению негативного влияния удаления тиомерсала на эффективность вакцины, необходимо выявление, по меньшей мере, одного наиболее зависимого от присутствия тиомерсала фактора (или тех факторов, для которых есть фармакологическое обоснование зависимости от наличия тиомерсала). Например, установлено, что нетипично высокая частота гипертермических реакций на введение одной вирусной вакцины, была связана с удалением из ее состава тиомерсала. Таким образом, ответственность заявителя заключается в представлении достоверных сведений по анализу безопасности и эффективности, полученных в исследованиях достаточной мощности.
29. Данные по безопасности комбинированной вакцины, содержащей консервант, альтернативный ртутьорганическому соединению не являются подтверждением безопасности вакцины не содержащей консервант, или содержащей его уменьшенное количество. В данном случае альтернативное соединение может выполнять все или некоторые функции тиомерсала. В таких случаях может потребоваться дополнительное исследование.
30. При этом данные, подтверждающие сохранность антигена в вакцине без тиомерсала, могут быть получены с учетом результатов исследования комбинированной вакцины без тиомерсала, в состав которой входит данный антиген.
7. Замена тиомерсала другими противомикробными средствами
31. Безопасность и эффективность лекарственного препарата могут быть косвенно подтверждены данными об иммуногенности вакцинного антигена в присутствии альтернативного консерванта: например, данные по влиянию 2-феноксиэтанола могут быть получены на основании клинических исследований лекарственного препарата, в котором вакцинный антиген, не содержащий консервант, был объединен в составе комбинированной вакцины с другими вакцинными антигенами в присутствии 2-феноксиэтанола. В этом случае результаты действительны только для вакцинных антигенов того же производителя. Аналогичные подходы к установлению относительной эффективности и безопасности после замены консерванта применяются и в случае удаления тиомерсала.
32. Положения настоящей главы устанавливают общие подходы для трех предлагаемых изменений: снижения концентрации, исключения или замены тиомерсала, однако каждая вакцина рассматривается уполномоченными органами (экспертными организациями) государств-членов при ее регистрации с учетом ее индивидуальных особенностей.
Глава 28. Требования к разработке и оценке качества вакцин для профилактики гриппа
1. Общие положения
1. Положения настоящей главы распространяются на процедуры, связанные с разработкой, качеством и оценкой качества сезонных, препандемических (зоонозных) и пандемических вакцин для профилактики гриппа, предназначенных для регистрации на таможенной территории Союза, а также при внесении изменений в регистрационное досье зарегистрированных сезонных, пандемических и препандемических (зоонозных) вакцин при изменении (обновлении) штаммового состава вакцин для профилактики гриппа.
2. Требования распространяются на инактивированные вакцины без адъюванта или с адъювантом, живые аттенуированные вакцины для профилактики гриппа (ЖГВ).
3. Положения настоящей главы также применимы к новым типам инактивированных вакцин, содержащих альтернативные вакцинные антигены или к живым вакцинам на основе новых генно-инженерных конструкций.
4. Требования к вакцинам на основе рекомбинантных генно-инженерных конструкций, совмещающих в одном продукте экспрессии эпитопы большого количества разных вирусов гриппа, и к вакцинам на основе нуклеиновых кислот не рассматриваются в настоящей главе.
5. По вопросам регистрации всех новых вакцин, к которым положения настоящей главы неприменимы в полной мере, заявителям следует обращаться за научной консультацией в уполномоченные органы (экспертные организации) государств-членов в соответствии со статьей 26 Правил регистрации и экспертизы.
6. Для целей настоящей главы понятия применяются в значениях, определенных приложением № 24 к Правилам регистрации и экспертизы.
2. Требования к качеству при подаче заявления о регистрации сезонных инактивированных вакцин для профилактики сезонного гриппа
7. К заявлению о регистрации новой сезонной вакцины для профилактики гриппа должен прилагаться комплект документов в соответствии с приложением № 1 к Правилам регистрации и экспертизы.
8. Сведения, касающиеся вопросов качества, должны быть представлены в модулях 2 и 3 регистрационного досье сезонных вакцин для профилактики гриппа.
9. Требования к модулю 3 регистрационного досье сезонных вакцин для профилактики гриппа, установленные пунктами 20 - 74 настоящей главы, приведены с указанием номеров разделов регистрационного досье в формате общего технического документа согласно приложению № 4 к Правилам регистрации и экспертизы.
10. В производстве вакцин для профилактики гриппа в качестве субстрата используют развивающиеся куриные эмбрионы (РКЭ) или подходящие культуры клеток, которые должны соответствовать требованиям общей фармакопейной статьи (монографии) Фармакопеи Союза, а при отсутствии в ней - требованиям общих фармакопейных статей (монографий) фармакопей государств-членов.
11. Вакцина для профилактики гриппа должна соответствовать требованиям фармакопейной статьи (монографии) Фармакопеи Союза, а при отсутствии в ней - требованиям фармакопейных статей (монографий) фармакопей государств-членов.
12. Развивающиеся куриные эмбрионы для посевного материала вируса гриппа должны поступать из изолированных, постоянно контролируемых хозяйств, свободных от специфической патогенной флоры (SPF).
13. Куриные эмбрионы, которые предстоит использовать для производства вакцины, должны быть взяты от здорового поголовья кур категории SPF или должны быть получены из птицехозяйств, благополучных по возбудителям, патогенным для человека (качество поставляемых эмбрионов должно быть подтверждено ветеринарными свидетельствами и входным контролем на отсутствие возбудителей, патогенных для человека (аденовирус, микоплазма, лейкоз птиц) или наличием документа производителя куриных эмбрионов, подтверждающего отсутствие возбудителей, патогенных для человека).
14. Данные, полученные при работе с различными штаммами, следует использовать для создания объединенной базы данных, которая используется, для того, чтобы детально описать требования к качеству вакцины после адаптации (оптимизации) процесса производства к определенному штамму при изменении (обновлении) штаммового состава сезонной вакцины.
2.1. Штаммы-кандидаты для производства сезонных вакцин для профилактики гриппа
15. Штаммы-кандидаты, используемые для получения сезонных вакцин для профилактики гриппа, представляют собой штаммы вируса гриппа, рекомендованные ВОЗ, и подходящие для производства сезонных вакцин. Проспективно, поставками таких штаммов производителям вакцин, для создания банков посевных материалов на производстве, занимаются сотрудничающие центры ВОЗ по гриппу (WHO Collaborating Centre (СС)), специализированные лаборатории ВОЗ (WHO Essential Regulatory Laboratory (ERL)) и другие сертифицированные лаборатории, специализирующиеся на получении штаммов-кандидатов для производства сезонных вакцин для профилактики гриппа.
16. Для того чтобы сделать процесс производства сезонных вакцин для профилактики гриппа более гибким в целях своевременного получения штамма-кандидата производители вакцин вправе разработать собственный штамм-кандидат, в случае, если такой штамм будет удовлетворять рекомендациям ВОЗ.
17. В связи с активной и непрерывной разработкой и модификацией методов и (или) технологий молекулярной биологии, необходимо обосновывать возможность их применения и пригодность для получения и аттестации штамма-кандидата для производства сезонной вакцины против гриппа. Производитель вакцины обязан установить пригодность штамма-кандидата для производства сезонной вакцины, а также создать посевной материал, соблюдая при этом рекомендации ВОЗ в отношении состава сезонной гриппозной вакцины.
Разработка штамма-кандидата для производства сезонных вакцин для профилактики гриппа
18. Штаммы-кандидаты для производства сезонной вакцины можно разработать с использованием одного из следующих субстратов:
развивающихся куриных эмбрионов;
клеток, полученных из развивающихся куриных эмбрионов;
клеток млекопитающих (в соответствии с указаниями Приложения № 1 к настоящей главе).
19. Штамм-кандидат для производства сезонной вакцины для профилактики гриппа может представлять собой:
высокопродуктивный реассортантный вирус, полученный с помощью метода классической реассортации. Этот вирус содержит кодирующие гемагглютинин (НА) и нейраминидазу (NA) сегменты генома от эпидемически актуального штамма вируса гриппа, рекомендованного ВОЗ, а оставшиеся сегменты - от высокорепродуктивного штамма-донора (PR8 или аналогичного штамма). Комбинация генома случайна, она определяется например как 5:3 или 6:2, где первое число означает количество сегментов генома высокорепродуктивного штамма, а второе - количество сегментов генома рекомендованного вируса гриппа дикого типа. Реассортант, как минимум, должен содержать гемагглютинин и нейраминидазу штамма вируса гриппа дикого типа. Сотрудничающий центр ВОЗ по гриппу, с применением установленной процедуры, должен подтвердить антигенное соответствие вируса-реассортанта штамму, рекомендованному ВОЗ;
реассортантный вирус, полученный с помощью методов обратной генетики (в том числе с использованием искусственно синтезированных последовательностей генов вируса гриппа). Данные штаммы конструируются с определенным сочетанием в геноме и, как правило, содержат кодирующие НА и NA сегменты генома от эпидемически актуального штамма вируса гриппа и оставшиеся шесть сегментов генома от PR8 или других подходящих высокопродуктивных донорских штаммов. Сотрудничающий центр ВОЗ по гриппу, с применением установленной процедуры, должен подтвердить антигенное соответствие вируса-реассортанта эпидемически актуальному штамму;
вирус, не являющийся реассортантным (вирус гриппа дикого типа).
Качество и оценка качества штамма-кандидата для производства сезонных вакцин для профилактики гриппа
20. Источник выделения и история пассажей штамма-кандидата, полученного в развивающихся куриных эмбрионах или культурах клеток должны быть известны и одобрены уполномоченными органами (экспертными организациями) государств-членов. Для штаммов-кандидатов, полученных производителем методом классической реассортации или обратной генетики, необходимо представить общее описание разработки штамма-кандидата для производства сезонной вакцины для профилактики гриппа (история разработки посевного материала, уровень пассажа). Соответствующие сведения включаются в раздел 3.2.S.2.3 модуля 3 регистрационного досье. Должны вестись лабораторные журналы, в которые необходимо включать документальное подтверждение, что во время проведения работ со штаммом-кандидатом, работы с любыми другими вирусами гриппа или генетическим материалом указанных вирусов не проводились в целях недопущения перекрестной контаминации. Необходимо учитывать положения приложения № 1 к настоящей главе.
21. В случае если в качестве штамма-кандидата будет использоваться реассортантный вирус, полученный с помощью методов обратной генетики (в том числе искусственно синтезированных генетических последовательностей вируса), необходимо учитывать требования к качеству, изложенные в пунктах 92 - 98 настоящей главы. В приложении № 2 к настоящей главе представлен пример схемы описания разработки штамма-кандидата для производства вакцины, полученного с помощью методов обратной генетики.
22. Если производитель самостоятельно разрабатывает реассортантный вирус (из вируса гриппа дикого типа или с использованием методов молекулярной биологии), необходимо проводить соответствующие испытания (изучение антигенных характеристик, анализ генетической последовательности). В том числе с использованием метода перекрестной реакции торможения гемагглютинации необходимо подтверждать антигенное соответствие вируса-реассортанта (то есть определение принадлежности его НА штамму дикого типа, рекомендованному ВОЗ). Оценку соответствия антигенной структуры полученного реассортанта также должен провести и документально подтвердить один из сотрудничающих центров ВОЗ по гриппу.
2.2. Оценка качества вирусного посевного материала
23. Сведения, касающиеся вопросов качества вирусного посевного материала: получение, аттестацию, испытания на присутствие посторонних агентов необходимо представить в разделе 3.2.S.2.3 модуля 3 регистрационного досье.
Получение вирусного посевного материала
24. Производство вакцины основано на использовании системы посевных вирусов (seedlot). Репродукцию посевного материала вируса гриппа осуществляют в развивающихся куриных эмбрионах или в подходящих культурах клеток, качество которых должно соответствовать требованиям общей фармакопейной статьи (монографии) Фармакопеи Союза, а при отсутствии в ней - требованиям общих фармакопейных статей (монографий) фармакопей государств-членов. Куриные эмбрионы, которые предстоит использовать, должны быть взяты от здорового поголовья кур категории SPF.
25. В соответствии с требованиями Правил производственной практики, указанные технологические процедуры следует проводить в асептических условиях с надлежащим микробиологическим контролем. Исходным штаммом для создания системы посевных вирусов является штамм-кандидат для производства вакцины, полученный из сотрудничающих центров ВОЗ по гриппу, специализированных лабораторий ВОЗ и других сертифицированных лабораторий, специализирующихся на получении штаммов-кандидатов для производства сезонных вакцин для профилактики гриппа, либо штамм-кандидат, разработанный производителем.
Контроль вирусного посевного материала
26. Антигены НА и NA каждого из посевных материалов проверяют на подлинность методами реакции торможения гемагглютинации (РТГА) и реакции ингибирования нейраминидазной активности (РИНА). Для этого используют специфичные антисыворотки, которые получают из сотрудничающих центров ВОЗ по гриппу. Если указанные реактивы недоступны или недостаточно специфичны, в отношении указанного показателя посевного вируса применяются альтернативные испытания (например, полимеразная цепная реакция (ПЦР)). Если подходящие реактивы есть в наличии, то для определения подлинности необходимо проводить соответствующие испытания.
27. Необходимо на уровне посевного вируса (рабочего посевного материала и (или) первого пассажа от рабочего посевного вируса (финальный вирусный сбор) проводить генетический анализ ответственных за синтез НА и NA генов каждого нового штамма вируса и сравнивать со штаммом-кандидатом для производства вакцины (или с общедоступной информацией из базы данных). Такую информацию по мере накопления опытных данных следует увязать с иммуногенностью и (или) эффективностью вакцины. Рабочей посевной материал должен быть не более 15 пассажей.
Испытания на присутствие посторонних агентов
28. Посевной вирус необходимо проверять на отсутствие посторонних агентов в соответствии с требованиями фармакопейных статей (монографий) Фармакопеи Союза, а при отсутствии в ней - с требованиями фармакопейных статей (монографий) фармакопей государств-членов на инактивированные вакцины для профилактики гриппа, полученные в развивающихся куриных эмбрионах или культурах клеток. Необходимо учитывать, что реактивы и субстраты животного происхождения, которые используются при приготовлении посевного материала для вакцины, потенциально могут представлять дополнительный риск вирусной безопасности.
29. Поскольку оценка на присутствие посторонних агентов в посевных вирусах, приготовленных с использованием развивающихся куриных эмбрионов или культур клеток из штаммов-кандидатов, разработанных с использованием методов классической реассортации или обратной генетики, будут отличаться, оценка риска должна быть проведена и включать в себя:
информацию о вирусах (новых и (или) появляющихся), которые потенциально могут присутствовать в клинических пробах (изолятах), используемых в производстве штаммов-кандидатов для вакцин. В качестве контаминирующих агентов допускается рассматривать такие патогенные микроорганизмы, как респираторно-синцитиальный вирус, аденовирус, вирус парагриппа, коронавирус, риновирус, энтеровирус, вирус Эпштейна-Барр, вирус простого герпеса, цитомегаловирус и микоплазмы;
восприимчивость к посторонним агентам субстратов для производства и выделения штамма;
риски вирусной безопасности, связанные с использованием при подготовке посевных вирусов сырья, реактивов и субстратов животного происхождения;
протоколы испытаний на посторонние агенты, которые проводились ранее.
30. Информацию, относящуюся к подготовке штамма-кандидата для производства сезонной вакцины, необходимо получить у поставщика штаммов-кандидатов для производства вакцины для профилактики гриппа. В случае непредставления такой информации, данный факт необходимо принимать во внимание при оценке рисков. Оценка рисков должна стать основанием для формирования спецификации и перечня испытаний посевных материалов на присутствие посторонних агентов. Оценка рисков пересматривается, когда становится доступной новая информация о потенциальных вирусных контаминантах, обоснование проводимых соответствующих испытаний представляется в рамках ежегодного изменения (обновления) штаммового состава.
31. Если субстрат оказывается восприимчив к контаминанту, обнаруженному в посевном материале, такой посевной материал считается непригодным для использования. Если субстрат к этому контаминанту устойчив, необходимо принять меры, чтобы убедиться, что в процессе производства контаминант в рабочем посевном материале элиминируется и (или) инактивируется.
32. В случае если необходимо прибегнуть к смене производственного штамма в сжатые сроки, производители вакцин для профилактики гриппа должны дополнительно разрабатывать методы, направленные на определение потенциальных контаминантов, патогенных для человека (например, мультиплексную ПЦР, которая может эффективно применяться в условиях временных ограничений производства сезонных вакцин).
33. При условии согласования с уполномоченными органами (экспертными организациями) государств-членов и после проведения валидации указанные методы применяются как альтернатива испытаниям на посторонние агенты, описанным в фармакопейной статье (монографии) Фармакопеи Союза, а при отсутствии в ней - описанным в фармакопеях государств-членов.
34. Допускается чтобы стратегии по обеспечению отсутствия посторонних агентов в готовой вакцине сочетали в себе испытания посевного вируса, соответствующие испытания на более поздних этапах производства (на уровне каждого из инактивированных моновалентных нерасфасованных продуктов), а также валидацию процессов производства. Выбранная стратегия должна быть должным образом обоснована для применяемой производственной платформы.
2.3. Оценка качества субстрата для культивирования вируса
35. При производстве вакцины каждый вирусный штамм выращивают в аллантоисной полости развивающихся куриных эмбрионов, или в диплоидной (перевиваемой) клеточной линии.
36. Развивающиеся куриные эмбрионы для посевного материала вируса гриппа должны поступать из изолированных, постоянно контролируемых хозяйств, свободных от специфической патогенной флоры SPF.
37. Куриные эмбрионы, которые предстоит использовать для производства вакцины, должны быть взяты от здорового поголовья кур категории SPF или получены из птицехозяйств, благополучных по возбудителям, патогенным для человека (качество поставляемых эмбрионов должно быть подтверждено ветеринарными свидетельствами и входным контролем на отсутствие возбудителей, патогенных для человека (аденовирус, микоплазма, лейкоз птиц) или наличием документа производителя куриных эмбрионов, подтверждающего отсутствие возбудителей, патогенных для человека).
Испытания на присутствие посторонних агентов
38. В дополнение к требованиям к клеточным субстратам, изложенным в настоящих Правилах и общих испытаний на посторонние агенты, клеточные субстраты для производства моновалентных нефасованных продуктов необходимо проверять на присутствие актуальных посторонних агентов (например, характерных для вида, который послужил источником клеток) или тех посторонних агентов, которые могли быть занесены с биологическими реактивами, используемыми при создании банка клеток.
39. Сведения о качестве субстрата, используемого для культивирования вируса включаются в раздел 3.2.S.2.3 модуля 3 регистрационного досье.
2.4. Разработка производственного процесса
40. В регистрационном досье необходимо представить подробное описание процесса производства, его контроль и разработку (разделы 3.2.S.2, 3.2.P.3, 3.2.Р.2.3 модуля 3 регистрационного досье).
41. Производственный процесс вакцин для профилактики гриппа может ежегодно разрабатываться или технически адаптироваться и (или) оптимизироваться в зависимости от специфических характеристик конкретного штамма вируса гриппа. Необходимо усовершенствовать процесс и механизм получения данных о продукте, опираясь на опыт производства в прошлом и новейшие исследования по изучению характеристик вакцины и процесса ее производства. Это позволяет лучше прогнозировать потенциальное влияние изменений процесса производства на качество вакцины. Опыт работы с различными штаммами допускается использовать для создания базы данных с целью получения детального представления о том, как действует ежегодная адаптация процессов производства для нового штамма, чтобы гарантировать в будущем, что внесение изменений в штаммовый состав не повлияет на качество вакцины.
42. В случае агрегации белка в активной фармацевтической субстанции (биологической субстанции) либо в вакцине допускается провести их оценку соответствующими методами анализа (например, с помощью метода динамического рассеяния света). В подобном случае, необходимо представить информацию об основной причине агрегации (например, характеристику, присущую штамму, влияние транспортировки, температуры, которые характерны для какого-то конкретного этапа и т. д.). Кроме того, необходимо описать соответствующую стратегию контроля. В дальнейшем необходимо обратить внимание на безопасность и иммуногенность вакцины, содержащей такие частицы.
2.5. Валидация производственного процесса
43. Требуется постоянно формировать валидационные данные, характеризующие производственный процесс, с целью подтверждения, что при установленных параметрах, процессы производства способны эффективно и воспроизводимо приводить к выпуску серий лекарственного препарата которые отвечают заданным в спецификации нормам и требованиям к качеству. Одним из важнейших этапов технологического процесса (критических точек производства) является инактивация вируса гриппа, используемого для производства вакцины. Процесс инактивации должен приводить к инактивации вируса гриппа в вакцине, не влияя на структуру антигенов НА и NA (не влияя на антигенные свойства вакцины). Исследования кинетики инактивации необходимо проводить с использованием трех производственных серий, изготовленных в промышленном масштабе.
44. В случае если это обосновано (например, когда подтверждена эквивалентность между процессом инактивации в промышленном и опытном масштабах производств), допускается использовать серии опытно-промышленного производства. Процесс инактивации должен быть валидирован.
45. Одним из важнейших этапов (критических точек производства), который необходимо включить в программу валидации процесса в отношении расщепленных или субъединичных вакцин, является также расщепление вируса гриппа. Эффективность расщепления необходимо подтверждать с помощью подходящих методов анализа. Необходимо суммировать данные о валидации процесса, как минимум, для трех последовательно произведенных серий.
46. Для аллантоисных вакцин или для вакцин, получаемых в культурах клеток необходимо подтвердить, что процесс инактивации способен инактивировать вирус лейкоза птиц и микоплазмы. Необходимо рассмотреть возможность подтверждения, инактивирует ли указанный процесс также и другие патогенные для птиц агенты (например, аденовирус птиц). Если условия инактивации модифицировались, необходимо представить уполномоченным органам (экспертным организациям) государств-членов данные, отражающие влияние этих изменений на способность инактивировать перечисленные возбудители, патогенные для птиц.
47. Этап инактивации вируса гриппа при производстве вакцины, а также и все другие этапы, связанные с инактивацией и (или) элиминацией контаминантов, необходимо оценивать на эффективность инактивации потенциальных контаминантов, которые могли быть привнесены на уровне посевного вируса.
48. Испытания эффективности инактивации вируса гриппа, для производства вакцины допускается проводить с использованием клеточного субстрата или любой другой клеточной системы, при условии, что проведено и валидировано испытание чувствительности указанных субстратов.
49. Описание, документацию и результаты исследований по валидации производственного процесса, критических точек производства или методов количественного определения, используемых в производственном процессе необходимо представить в разделах 3.2.S.2.5 и 3.2.Р.5.3 модуля 3 регистрационного досье.
2.6. Описание характеристик вакцины
50. Поскольку определенные характеристики вакцины могут присутствовать только у определенных штаммов возбудителя, проведение расширенных исследований позволяет охарактеризовать процесс производства и вакцину, а также представить информацию о сохранении свойств вакцины от сезона к сезону. Такое изучение позволит установить соответствующие нормы спецификации и подтвердить научную оценку сопоставимости после того, как в вакцину или в процесс ее производства будут внесены изменения.
51. Вид необходимых исследований для изучения определенных характеристик штамма, зависит от типа вакцины (например, цельновирионная, расщепленная или субъединичная).
52. Необходимо изучить и охарактеризовать содержащиеся в активной фармацевтической субстанции (биологической субстанции) действующие вещества и технологические примеси.
53. Биологические, иммунологические и физико-химические свойства антигена НА необходимо подтверждать с помощью широкого спектра новейших методов анализа (для изучения химических, физических и биологических характеристик НА применяются следующие методы: определение титра гемагглютинина, реакция торможения гемагглютинации, вестерн-блоттинг, картирование эпитопов, определение иммуногенности на мышах, инфицирование иммунизированных хорьков, электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-PAGE), масс спектрометрия с матричной лазерной десорбцией/ионизацией (MALDI/MS), ВЭЖХ (HPLC), просвечивающая электронная микроскопия, изопикническое ультрацентрифугирование в градиенте плотности, динамическое рассеяние света, триптическое пептидное картирование, секвенирование аминокислотной последовательности). Присутствие и тип антигена NA необходимо подтверждать с использованием адекватных ферментативных или иммунологических методов на моновалентных объединенных вирусных сборах. Необходимо уделить особое внимание характеристике и количественной оценке антигенов (кроме НА), которые могут влиять на иммуногенность вакцины (в случае если такая характеристика технически осуществима).
54. Допускаются с учетом развития науки и биотехнологии использование новых технологий и модификация существующих в рамках внесения изменений в регистрационное досье вакцины, не связанных с процедурами ежегодного обновления штаммового состава.
55. В случае присутствия в активной фармацевтической субстанции и (или) вакцине агрегированных частиц, необходимо исследовать размер состав, количественное содержание и профиль растворения этих частиц и учесть безопасность и иммуногенность вакцины, содержащей агрегированные частицы.
56. Необходимо идентифицировать и количественно определить технологические примеси (например, овальбумин в аллантоисных вакцинах или белки клетки-хозяина, остаточная ДНК клетки-хозяина в вакцинах, полученных в культуре клеток, примеси появившиеся на дальнейших этапах производства, например, реактивы, используемые для инактивации и (или) элиминации), и затем использовать эти данные для разработки нормативных требований, отраженных в спецификации при выпуске.
57. Соответствующие сведения необходимо включать в разделы 3.2.S.3.1, 3.2.S.3.2, 3.2.S.4.1 - 3.2.S.4.5 модуля 3 регистрационного досье.
58. Если метод производства вакцины, получаемой в культуре клеток, валидирован (с использованием широкого спектра штаммов вируса гриппа, чтобы продемонстрировать соответствующее сокращение остаточной ДНК и (или) белков клетки-хозяина), проводимое в штатном порядке испытание на остаточную ДНК и белки клетки-хозяина допускается не проводить.
2.7. Форма выпуска
59. Разработка специализированной формы выпуска для определенной целевой популяции должна быть обоснована данными о качестве, включая, данные о сопоставимости, валидации производственного процесса и данные о стабильности вакцины.
2.8. Стандартизация вакцин
60. Основным методом определения специфической активности инактивированных сезонных вакцин для профилактики гриппа, является определение количества гемагглютинина с помощью реакции одиночной радиальной иммунодиффузии (ОРИД). Достоверной корреляции между специфической активностью вакцины и клинической эффективностью не существует, поскольку эффективность зачастую зависит от типа вакцины (например, цельновирионная, расщепленная или субъединичная), состава (например, наличие адъюванта), производственных процессов, пути введения вакцины, совпадения вакцинного штамма с циркулирующим доминантным штаммом вируса гриппа дикого типа, вариабельности серологических методов количественного определения, недостаточности объективных данных об истинных серологических коррелятах защиты. Проведение реакции одиночной радиальной иммунодиффузии является подтверждением того, что антигенная специфичность и содержание антигенов НА в вакцине остается надлежащим и не меняется от серии к серии. Для дозы инактивированной сезонной вакцины априорно установлено, что содержание НА в вакцине, определяемое в реакции одиночной радиальной иммунодиффузии, составляет 15 мкг для каждого штамма, входящего в состав вакцины.
61. Необходимо представить информацию о таких показателях качества как количество, антигенная специфичность соответствующих антигенов гриппа, и о составе вакцины (например, добавлен адъювант или нет). Влияние этих факторов на иммуногенность, профилактическую эффективность и безопасность вакцины необходимо фиксировать и изучать.
62. Для реакции одиночной радиальной иммунодиффузии требуются штаммоспецифичные реактивы, а сроки их получения могут задержать выпуск первых серий вакцины. В этой связи необходимо использовать альтернативные методы количественного определения (иммуноферментный анализ, ВЭЖХ и т. д.), которые применяются, пока реактивы для реакции одиночной радиальной иммунодиффузии недоступны.
63. Исходя из этого, производителям вакцин необходимо искать альтернативные методы, а также улучшать метод количественного определения специфической активности с помощью реакции одиночной радиальной иммунодиффузии, сотрудничая при этом с регуляторными и научными лабораториями. Особое внимание необходимо уделять методам, которые позволяют определять, с точки зрения биологической значимости, меру специфической активности (количество функционально активного белка) и (или) могут указывать на стабильность. Поскольку добиться полной идентичности для методов определения специфической активности вакцин невозможно, результат считается оптимальным если значения специфической активности, полученные с помощью альтернативного метода количественного определения, будут соответствовать значениям, полученным с помощью реакции одиночной радиальной иммунодиффузии. Производителям необходимо обосновать и валидировать использование таких методов количественного определения НА для внутрипроизводственных контрольных испытаний и испытаний при выпуске (до или после того, как станут доступны реактивы для реакции одиночной радиальной иммунодиффузии и (или) в качестве исследований стабильности. Это критически важно в тех случаях, когда требуются отдельные методы определения подлинности, с одной стороны, и методы анализа для внутрипроизводственного контроля (анализа выпускаемой серии (партии), оценки стабильности), с другой стороны. Стратегия использования независимого от антител альтернативного метода анализа должна учитывать то, как будет гарантироваться иммуногенность антигена (например, специфичность, антигенность) и сохранение показателей иммуногенности от серии к серии.
64. Обоснованность, пользу и применимость альтернативных методов анализа для количественного определения специфической активности вакцины необходимо в последующем оценивать в ходе процесса валидации. В случае если предполагается применить для испытаний при выпуске серии альтернативный метод, необходимо проводить сравнение между количественным определением альтернативным методом и количественным определением с помощью реакции одиночной радиальной иммунодиффузии, используя при этом несколько штаммов. Вышеуказанное важно при принятии решения о стратегии использования альтернативного метода, в случае, когда реагенты для одиночной радиальной иммунодиффузии уже будут доступны.
65. При оценке специфической активности культуральных вакцин, в производстве которых использовался вирус-реассортант, полученный пассированием в развивающихся куриных эмбрионах, для постановки метода реакции одиночной радиальной иммунодиффузии допускается применение стандартных образцов, полученных с использованием развивающихся куриных эмбрионов. Исследования оценки необходимости в стандартных образцах, произведенных в культуре клеток, в которых параллельно используют и аллантоисные стандартные образцы для реакции одиночной радиальной иммунодиффузии (антигены и антисыворотки) в настоящее время признаются непригодными для проведения.
66. Соответствующие сведения необходимо включать в разделы 3.2.Р.5.1 - 3.2.Р.5.6 и 3.2.Р.6 модуля 3 регистрационного досье.
2.9. Оценка адъювантов
67. При использовании адъювантов, по вопросам качества, необходимо учитывать требования главы 16 настоящих Правил. Регистрационное досье должно содержать следующую подробную информацию об адъюванте:
происхождение или источник исходных материалов;
процесс производства;
физические и химические характеристики;
результаты контрольных испытаний и исследований стабильности;
взаимодействие с вакцинным антигеном.
68. Необходимо подробно описать этапы разработки технологического процесса производства адъюванта.
69. В соответствующих случаях представляется детальная информация о необходимости немедленного смешения адъюванта с антигеном. Необходимо учесть действие, оказываемое длительностью смешения на важнейшие характеристики антигена (адъюванта) и их комбинации. Необходимо соответствующим образом валидировать процесс хранения в течение предполагаемого срока годности (срока хранения) после вскрытия.
70. Необходимо представить в регистрационном досье данные, касающиеся использования многодозовых упаковок (например, многократного прокалывания пробок многодозовых упаковок для набора 1 дозы, правильного подбора калибра и длины иглы для набора и введения вакцины). Описание внешнего вида каждого из компонентов должно быть достаточно подробным, в том числе должна быть включена информация относительно допустимости применения вакцины в случаях, когда в готовом лекарственном препарате присутствуют видимые частицы (агрегаты). В случае если предполагается наличие допустимых излишков, возникших при наполнении контейнеров с вакциной, они должны быть адекватно обоснованы и учтены при написании инструкций по смешиванию (применению) вакцины.
71. Сведения об адъюванте необходимо включать в разделы 3.2.Р.1 - 3.2.Р.8 модуля 3 регистрационного досье вакцины.
2.10. Исследование стабильности и срок годности (срок хранения)
72. Данные о стабильности вакцины для профилактики гриппа необходимо получать в соответствии с требованиями Фармакопеи Союза, а при отсутствии в ней - с требованиями общих фармакопейных статей (монографий) фармакопей государств-членов и главы 8 настоящих Правил. Необходимо представлять данные о стабильности, включая результаты исследований естественного, ускоренного хранения и (или) стресс-исследований, для активной фармацевтической субстанции (моновалентной субстанции) и для вакцины (готовой лекарственной формы), чтобы подтвердить установленные максимальный срок годности (срок хранения) активной фармацевтической субстанции и срок годности (срок хранения) вакцины, соответственно.
73. Необходимо разработать протокол исследований стабильности вакцины. Должна быть описана и обоснована процедура определения срока годности (срока хранения) и даты истечения срока годности (срока хранения).
74. Соответствующие сведения необходимо представить в разделах 3.2.S.7.1 - 3.2.S.7.3 и 3.2.Р.8.1 - 3.2.Р.8.3 модуля 3 регистрационного досье.
3. Требования к качеству при подаче заявления о внесении изменений в штаммовый состав сезонных инактивированных вакцин для профилактики сезонного гриппа
75. ВОЗ дважды в год (как правило, в феврале - марте для Северного полушария и в сентябре для Южного полушария) обновляет свои рекомендации в отношении штаммов вируса гриппа А и В, которые следует использовать в производстве сезонных вакцин для профилактики гриппа, предназначенных для предстоящего эпидемического сезона.
76. В рамках процедуры внесения изменений в штаммовый состав заявителю допускается вносить изменения, касающиеся только изменения (обновления) штаммового состава. В рамках процедуры внесения изменений в штаммовый состав уполномоченные органы (экспертные организации) референтного государства и государств признания не вправе требовать от заявителя внесений каких-либо иных изменений в состав регистрационного досье вакцины для профилактики гриппа, за исключением изменений, обусловленных изменением ее штаммового состава.
77. Заявители при подаче заявления о внесении изменений в штаммовый состав сезонных инактивированных вакцин для профилактики гриппа представляют комплект документов в соответствии с приложением № 24 к Правилам регистрации и экспертизы.
3.1. Качество и оценка качества штамма-кандидата для производства сезонной вакцины
78. Применимы требования, изложенные в пунктах 15 - 22 настоящей главы.
3.2. Качество и оценка качества вирусного посевного материала
79. Применимы требования, изложенные в пунктах 23 - 34 настоящей главы.
80. Необходимо представить обновленные данные по оценке рисков потенциальной контаминации посторонними агентами, в том числе информацию о новых и (или) вновь возникающих вирусах, которые потенциально могли бы присутствовать в клинических пробах (изолятах), которые использовались для получения штамма-кандидата, предназначенного для производства сезонной вакцины. Если посевной материал проверяется на наличие посторонних агентов с помощью полимеразной цепной реакции, и, если уполномоченным органом (экспертной организацией) референтного государства была согласована необходимость проведения дополнительных ПЦР-испытаний посевного материала, эти данные необходимо включить в раздел 3.2.S.2.3 регистрационного досье.
81. Для создания базы данных о качестве сезонных вакцин для профилактики гриппа проводится генетический анализ генов НА и NA для каждого нового штамма вируса гриппа на этапе посевного материала (рабочего посевного материала и (или) первого пассажа от рабочего посевного вируса (финальный вирусный сбор)) и сравнение с данными анализа производственного штамма-кандидата или материалами с открытых баз данных. Полученные данные таких анализов перспективно (после получения большего массива данных) следует связать с иммуногенностью (профилактической эффективностью вакцины). Результаты генетического анализа представляются в рамках ежегодного обновления штаммового состава или уже после него. Необходимо представить данные до выбора штаммов используемых в следующей кампании иммунизации против гриппа.
3.3. Разработка производственного процесса
82. Изменения в производственном процессе вакцины, связанные с оптимизацией технологических процессов, обусловленные специфическими характеристиками штамма должны быть обоснованы, соответствующие сведения необходимо включить в раздел 3.2.S.2.6 модуля 3 регистрационного досье. Изменения в описание производственного процесса и указание требований к контролю качества, в процессе производства подобранные специально под штамм вносятся при необходимости. Соответствующие сведения необходимо включать в разделы 3.2.S.2.2, 3.2.S.2.4 модуля 3 регистрационного досье.
83. Необходимо представить в разделе 3.2.Р.3.2 модуля 3 регистрационного досье подробную производственную рецептуру (состав на серию) в связи с изменением (обновлением) штаммового состава. Описание лекарственного препарата и его состав в включаются в раздел 3.2.Р.1 модуля 3 регистрационного досье.
3.4. Валидация производственного процесса
84. При изменении (обновлении) штаммового состава сезонных вакцин для профилактики гриппа необходимо представить результаты исследований по валидации для критических точек производства в разделе 3.2.S.2.5 модуля 3 регистрационного досье. Необходимо обосновать адекватность способов инактивации и в случаях когда применимо, эффективность расщепления вируса.
85. Результаты анализа серии первых 3 моновалентных нефасованных продуктов (включая показатели подлинности гемагглютинина и нейраминидазы), полученных из рабочего посевного материала, должны быть представлены в разделе 3.2.S.4.4 модуля 3 регистрационного досье.
3.5. Описание характеристик вакцины
86. В силу временных ограничений при ежегодном изменении (обновлении) штаммового состава вакцин для профилактики гриппа расширенное изучение характеристик активной фармацевтической субстанции нецелесообразно, однако эти данные могут способствовать лучшему пониманию процессов производства и продукции и дать информацию о постоянстве характеристик вакцины от сезона к сезону. При подаче заявления о внесении изменений в штаммовый состав сезонных инактивированных вакцин для профилактики гриппа необходимо описать в модуле 3 регистрационного досье выборочно несколько исследований. Эти исследования, в том числе, включают в себя информацию о распределении частиц по размерам, наличии агрегатов. Соответствующие сведения включают в раздел 3.2.S.3 модуля 3 регистрационного досье.
3.6. Стандартизация вакцины
87. Необходимо представить результаты валидации аналитических методик, на изменение которых может повлиять замена одного или нескольких штаммов (например, валидация метода реакции одиночной радиальной иммунодиффузии для нового штамма (штаммов) с учетом соответствующих стандартных образцов). Данные о валидации необходимо представить в разделе 3.2.S.4.3 для моновалентных нефасованных продуктов, а также трехвалентных нерасфасованных продуктов или вакцин в разделе 3.2.Р.5.3 модуля 3 регистрационного досье. При проведении повторной валидации аналитической методики испытания методом реакции одиночной радиальной иммунодиффузии, необходимо включать данные об аттестации сыворотки, подлинности и (или) специфичности, точности, прецизионности (внутрилабораторной прецизионности, воспроизводимости), линейности и (или) диапазоне применения и робастности.
88. Необходимо представить в виде таблиц копии утвержденных спецификаций для моновалентных нефасованных продуктов в разделе 3.2.S.4.1 модуля 3 регистрационного досье и для лекарственного препарата в разделе 3.2.Р.5.1 модуля 3 регистрационного досье, а также обзор аналитических методик в разделе 3.2.Р.5.2 модуля 3 регистрационного досье.
3.7. Исследование стабильности и срок годности (срок хранения)
89. Результаты исследований стабильности моновалентных нефасованных продуктов при хранении в условиях реального времени и «ускоренного старения» представляют с целью подтверждения максимального срока хранения, а также такие исследования могут послужить дополнительным доказательством качества вакцины с различным штаммовым составом. Поэтому при изменении (обновлении) штаммового состава проводятся исследования стабильности для каждого нового штамма, чтобы накопить базу данных о качестве вакцины. Необходимо представить результаты исследований стабильности моновалентных нерасфасованных продуктов, если предполагается их использование более 1 года. Соответствующие сведения включают в раздел 3.2.S.7 модуля 3 регистрационного досье.
90. Для готовой вакцины следует представлять в разделе 3.2.Р.8 модуля 3 регистрационного досье данные о стабильности как за предыдущий сезон, так и обязательство продолжать исследования стабильности нескольких серий вакцины по программе, описанной в протоколе пострегистрационных исследований стабильности.
3.8. Дополнительные данные по качеству вакцины
91. В случае необходимости, заявитель вправе представить обновление или дополнение предыдущей версии резюме по качеству модуля 2 регистрационного досье.
4. Требования к качеству при подаче заявления о регистрации препандемических (зоонозных) вакцин для профилактики гриппа
4.1. Штаммы-кандидаты для производства препандемических (зоонозных) вакцин для профилактики гриппа
92. Применимы требования, изложенные в пунктах 15 - 22 настоящей главы.
93. Заявителям необходимо обосновать выбор штамма-кандидата. В данном случае, приводится ссылка на опубликованную ВОЗ информацию, касающуюся антигенных и генетических характеристик A(H5N1), A(H7N3), A(H7N9), A(H9N2) и вариантных штаммов вируса гриппа и штаммов-кандидатов, разработанных для возможного использования в составе препандемических (зоонозных) вакцин для профилактики гриппа.
94. В случае, если штамм-кандидат для производства препандемической (зоонозной) вакцины разрабатывается на основе высокопатогенного вируса гриппа птиц подтипа (серотипа) Н5 или Н7 или от подобных вирусов, необходимо с помощью испытаний in vitro и in vivo подтвердить элиминацию высокопатогенного фенотипа гриппа птиц. Если серотип вируса гриппа не обладает высокой патогенностью, то такие испытания включают аналогичные проводимым для штаммов-кандидатов для производства вакцин против сезонного гриппа испытания на определение антигенных характеристик и анализ генетических последовательностей, также дополнительно включается оценка профиля безопасности.
95. Пандемический штамм-кандидат (наиболее вероятно птичьего, свиного или человеческого происхождения) выделяется с помощью одной из процедур, описанных в пунктах 15 - 22 настоящей главы. Однако, допускается использование нереассортантного вируса дикого типа (высоко- или низкопатогенного).
96. Примеры подходящих вирусов для использования в качестве штаммов-кандидатов для производства зоонозных вакцин, включают в себя:
реассортантный вирус подтипа H5N1, полученный из высокопатогенного штамма с помощью методов обратной генетики. Список доступных штаммов-кандидатов подтипа H5N1 для производства препандемических (зоонозных) представлен на сайте ВОЗ. Ввиду того, что наиболее часто встречается заражение человека вирусом Н5, преимущество выбора этого подтипа заключается в том, что штамм является потенциально пандемическим и производится с помощью методов обратной генетики, наиболее многообещающего метода разработки штаммов-кандидатов для пандемических вакцин из высокопатогенных подтипов вируса гриппа Н5 или Н7;
реассортантный вирус подтипа H7N1, полученный из высокопатогенного вируса гриппа птиц с помощью методов обратной генетики. С вирусами подтипов H7N1 и H7N7 связывают вспышки гриппа у сельскохозяйственных птиц, в том числе, вирусы подтипа H7N7 ассоциируются с заражением человека;
вирус гриппа птиц подтипа H5N3. Вакцины, полученные из штамма A/Duck/Singapore/97 подтипа H5N3, уже прошли клинические исследования. Этот штамм близок в антигенном отношении к высокопатогенному штамму A/Hong Kong/156/97 подтипа H5N1. Допускается возможность использования других низкопатогенных подтипов Н5;
вирус подтипа H7N9. Вирус гриппа птиц A(H7N9) - подтип вируса гриппа, который был обнаружен у птиц. С марта 2013 года выявлены случаи заражения как птиц, так и людей. Заболевание вызывает большую обеспокоенность, поскольку у большинства пациентов протекает тяжело. Для разработки вакцин против A(H7N9) с целью обеспечения готовности к возможной пандемии рекомендованы вирусы, подобные A/Anhui/1/2013 (A/Shanghai/2/2013 - вирус, подобный A/Anhui/1/2013);
вирус подтипа H9N2. Характерные для человека вирусы подтипа H9N2, такие как штамм A/Hong Kong/1073/99, уже используются для производства экспериментальных вакцин и прошли клинические исследования. Продолжают поступать сообщения о единичных случаях заражения людей вирусом подтипа H9N2. Есть предварительные доказательства, что лица, рожденные до 1968 года, могут быть праймированы, что повышает иммуногенность вакцин против H9N2. Поэтому необходимо рассмотреть вопрос деления субъектов клинических исследований вакцин против вируса подтипа H9N2 на группы по возрасту;
вирус гриппа человека подтипа H2N2. Штамм A/Singapore/1/57 вызвал пандемию в 1957 году и в последнее время используется в производстве новых экспериментальных вакцин. При проведении клинических исследований вакцин против вируса подтипа H2N2 необходимо учитывать праймированность лиц, родившихся до 1968 года.
97. Если подготовка штамма-кандидата для производства препандемической (зоонозной) вакцины предполагает использование методов обратной генетики (в том числе искусственно синтезированных последовательностей генов вируса гриппа) для генетической модификации вируса и последующей репродукции в клетках млекопитающих, необходимо проанализировать большее количество параметров, касающихся качества, чем при подготовке штамма-кандидата методом классической реассортации.
98. Использование обратной генетики требует применения клеток млекопитающих при разработке штамма-кандидата для производства вакцины, что влечет за собой дополнительные требования для обеспечения безопасности и качества вакцины. Использование клеток млекопитающих при разработке производственного штамма-кандидата для вакцины с помощью методов обратной генетики требует соблюдения минимального набора параметров:
клеточный субстрат, используемый для разработки производственного штамма-кандидата для вакцины, должен отвечать требованиям, изложенным в соответствующей статье Фармакопеи Союза, а при отсутствии в ней - требованиям общих фармакопейных статей (монографий) фармакопей государств-членов;
клеточные субстраты, предназначенные для производства вакцин для медицинского применения должны относиться к банку клеток, одобренному для производства вакцин для медицинского применения уполномоченными органами (экспертными организациями) государств-членов;
исходные материалы, применяемые при разработке штамма-кандидата для производства препандемической (зоонозной) вакцины с помощью обратной генетики должны соответствовать требованиям по обеспечению безопасности при использовании бычьей сыворотки и требованиям Фармакопеи Союза по минимизации риска передачи возбудителей губчатой энцефалопатии животных, а при отсутствии в ней - требованиям по минимизации риска передачи возбудителей губчатой энцефалопатии животных фармакопей государств-членов;
исходные материалы, используемые при разработке штамма-кандидата с помощью обратной генетики, могут потенциально оказать влияние на безопасность вакцины с точки зрения контаминации вирусами, бактериями, грибами и микоплазмами. Возможные риски по безопасности, связанные с исходными материалами, необходимо учитывать заявителем при проведении общей оценки безопасности вакцины.
4.2. Качество и оценка качества вирусного посевного материала
99. Применимы требования, изложенные в пунктах 23 - 34 настоящей главы.
100. В случае если в производстве вакцины используется генетически модифицированный с целью снижения патогенности штамм-кандидат определенных серотипов Н5 и Н7 или штаммы-кандидаты, полученные на основе низкопатогенных вариантов Н5 и Н7, необходимо подтвердить, что последовательность гемагглютинина в сайте протеолитического расщепления на этапе посевного вирусного материала идентична последовательности штамма-кандидата в целях обоснования сохранения свойств низкой патогенности (то есть отсутствует удлинение цепи полиосновных аминокислот в сайте расщепления гемагглютинина).
101. Подобные исследования необходимо также проводить для трех серий вирусного посевного материала на уровне пассажа для производства готовой вакцины. Допускается сотрудничество производителей с ВОЗ, сотрудничающими и контрольными лабораториями ВОЗ, аккредитованными государственными референс-центрами (когда это возможно), для совместного изучения характеристик посевного материала (например, его подлинности, гемагглютинирующего титра, молекулярных и генетических свойств).
4.3. Качество и оценка качества субстрата для культивирования вируса
102. Применимы требования, изложенные в пунктах 35 - 37 настоящей главы.
4.4. Разработка производственного процесса
103. Производство препандемической (зоонозной) вакцины против гриппа должно осуществляться по ранее утвержденному для производства сезонной вакцины производственному процессу, либо по новому разработанному производственному процессу. В любом из случаев разработку производственного процесса необходимо подробно описать в регистрационном досье. Для каждого из новых вновь разработанных производственных процессов требуется больший объем информации, чем для ранее валидированных производственных процессов. Процесс производства препандемической (зоонозной) вакцины должен быть технически адаптирован и соответствовать требованиям к производству вакцины в условиях предпандемии.
104. Оптимизация технологических условий должна быть обоснована и валидирована. Необходимо получить детальные данные о вакцинах и процессах производства, основанные на прошлом опыте производства аналогичных вакцин, исследованиях современных процессов производства и изучении характеристик новых вакцин. Потенциальное влияние изменений процессов производства на качество вакцины необходимо проверить по критическим показателям качества, основываясь на исследованиях, проведенных при разработке препандемических (зоонозных) и сезонных вакцин. Данные, полученные при работе с различными препандемическими и сезонными штаммами, допускается использовать для создания базы данных, исмпользуемой для более подробного описания требований к качеству вакцины при внесении изменений в штаммовый состав во время выбора зоонозных штаммов в условиях предпандемии.
4.5. Валидация производственного процесса
105. Применимы требования, изложенные в пунктах 43 - 49 настоящей главы.
4.6. Описание характеристик вакцины
106. Применимы требования, изложенные в пунктах 50 - 58 настоящей главы.
4.7. Форма выпуска
107. Допускается выпускать препандемические (зоонозные) вакцины для профилактики гриппа как в многодозовой, так и в однодозовой упаковке.
108. При использовании многодозовых упаковок проводится оценка необходимости добавления антимикробных консервантов, учитывая возможность контаминации в ходе применения и максимальный рекомендуемый срок хранения после вскрытия. В соответствующих случаях проводятся испытания вакцины на антимикробные консерванты в нефасованном виде. Заявителю необходимо выяснить, являются ли антимикробные консерванты мешающим фактором проведения других испытаний.
109. Если в состав вакцины против гриппа входит тиомерсал, заявитель должен обосновать конечное содержание тиомерсала.
110. Рекомендуемый срок хранения после вскрытия упаковки следует валидировать. Необходимо уделить особое внимание возможности потенциальной контаминации вакцины в многодозовой упаковке, связанной с неоднократным прокалыванием пробок многоразовых емкостей (например, закупорка просвета иглы кусочком пробки, выбор калибра и длины игл для набора и введения вакцины). Для многодозовых упаковок, используемых для вакцинации детей и взрослых, необходимо валидировать излишки объема содержимого, чтобы гарантировать, что вакцины достаточно для введения максимального количества детских доз.
111. В случае необходимости разработки вакцины в определенной форме выпуска, состав которой отличается от ранее зарегистрированной вакцины с целью применения такой новой вакцины у определенной целевой популяции (например, детская вакцина, содержащая половину дозы антигена с полной дозой адъюванта, или любую другую комбинацию) или в случае, когда необходимо помещение вакцины в другую первичную упаковку, разработка вакцины должна быть обоснована и подтверждена соответствующими данными о качестве, содержащими информацию о совместимости компонентов вакцины и упаковки, валидации производственного процесса и стабильности.
4.8. Стандартизация вакцины
112. Применимы требования, изложенные в пунктах 60 - 66 настоящей главы.
4.9. Оценка адъювантов
113. Применимы требования, изложенные в пунктах 67 - 71 настоящей главы.
4.10. Исследование стабильности и срок годности (срок хранения)
114. Необходимо представить данные о стабильности, включающие в себя данные исследования естественного, ускоренного хранения и (или) стресс-исследования для активной фармацевтической субстанции (моновалентного нерасфасованного продукта) и вакцины в готовой лекарственной форме, чтобы подтвердить максимальный срок годности (срок хранения) соответственно.
115. При подаче заявления о регистрации в регистрационное досье необходимо включать данные о стабильности в реальных условиях хранения в течение 6 месяцев. Продлевать срок годности до 1 года необходимо на основании данных исследования стабильности в реальном времени.
Исследования стабильности после вскрытия упаковки
116. Необходимо представить результаты исследований стабильности и изучения характеристик вакцины. Для подтверждения соответствия вакцины спецификации по критическим показателям качества (таким как параметры, указывающие на стабильность и микробиологическую чистоту), необходимо использовать современные методы. Исследования стабильности лекарственного препарата после вскрытия упаковки должны проводиться в условиях, имитирующих реальное извлечение доз из многодозового флакона, так чтобы пробка прокалывалась столько раз, сколько этого требует максимальное количество извлечений в естественных условиях.
5. Требования к качеству при подаче заявления о внесении изменений в штаммовый состав препандемических (зоонозных) вакцин для профилактики гриппа
117. В случае необходимости заявители могут подать заявление об изменении штаммового состава зарегистрированной препандемической (зоонозной) вакцины. Причинами внесения изменений в штаммовый состав препандемической вакцины являются: низкая или незначительная перекрестная реактивность и перекрестная протективность к дрейфовым вариантам вируса гриппа и (или) наличие заключения эпидемиологической службы государства-члена о том, что с наибольшей долей вероятности пандемия будет вызвана вирусом гриппа (внутри клайды или подклайда) с альтернативным подтипом гемагглютинина.
118. Требования к объему представляемых данных о качестве зависят от вида изменения штамма:
а) штамм, входящий в состав зарегистрированной вакцины, заменяется другим штаммом того же самого подтипа (например, исходный штамм подтипа H5N1 другим штаммом подтипа H5N1). В этом случае держателю регистрационного удостоверения необходимо представить все данные о производстве и качестве, относящиеся к новому штамму. Требуемая информация аналогична информации, необходимой для подачи заявления на внесение изменений в штаммовый состав сезонной вакцины;
б) штамм, входящий в состав зарегистрированной вакцины, заменяется штаммом с другим подтипом гемагглютинина и (или) нейраминидазы (например, исходный штамм подтипа H5N1 заменяется штаммом подтипа H7N9). В случае таких изменений штаммового состава заявителю вправе получить консультацию уполномоченных органов (экспертных организаций) государств-членов об объеме представляемых данных, поскольку имеется вероятность дополнительного проведения доклинических и клинических исследований, или отдельного представления их результатов (при их наличии).
6. Требования к качеству при подаче заявления о регистрации пандемических вакцин для профилактики гриппа до признания пандемической ситуации (вакцины готовности к пандемии)
119. Как и в случае с вакцинами против сезонного гриппа, большинство пандемических вакцин производится с использованием развивающихся куриных эмбрионов, либо клеточного субстрата. Пандемическая вакцина против гриппа и клеточный субстрат, используемый для разработки производственного штамма-кандидата для этой вакцины должны соответствовать требованиям Фармакопеи Союза, а при отсутствии в ней - требованиям фармакопей государств-членов на инактивированные вакцины против гриппа, полученные с использованием развивающихся куриных эмбрионов или культур клеток, в зависимости от конкретного случая.
6.1. Подача заявления о регистрации вакцин готовности к пандемии
120. В целях подготовки к пандемии производители вакцин вправе подать в уполномоченный орган (экспертную организацию) государств-членов заявление о регистрации кандидатной пандемической вакцины, содержащей штамм вируса с пандемическим потенциалом (вакцину готовности к пандемии).
121. В случае угрозы развития пандемической ситуации регистрационное досье вакцины готовности к пандемии должно содержать данные о потенциальном пандемическом штамме (штаммах) в соответствии с настоящей главой и приложением № 24 к Правилам регистрации и экспертизы.
6.2. Штаммы-кандидаты для производства вакцин готовности к пандемии
122. Применимы требования, изложенные в пунктах 15 - 22 и 92 - 98 настоящей главы.
6.3. Качество и оценка качества вирусного посевного материала
123. Применимы требования, изложенные в пунктах 23 - 34 и 99 - 104 настоящей главы.
6.4. Качество и оценка качества субстрата для культивирования вируса
124. Применимы требования, изложенные в пунктах 35 - 37 настоящей главы.
6.5. Разработка производственного процесса
125. Применимы требования, изложенные в пунктах 103 и 104 настоящей главы.
6.6. Валидация производственного процесса
126. Применимы требования, изложенные в пунктах 43 - 49 настоящей главы.
6.7. Описание характеристик вакцины
127. Применимы требования, изложенные в пунктах 50 - 58 настоящей главы.
6.8. Форма выпуска
128. Допускается выпуск вакцин готовности к пандемии для профилактики пандемического гриппа как в многодозовой, так и в однодозовой упаковке.
129. К форме выпуска вакцин готовности к пандемии применимы требования, изложенные в пунктах 107 - 111 настоящей главы.
6.9. Стандартизация вакцины
130. Применимы требования, изложенные в пунктах 60 - 66 настоящей главы.
6.10. Оценка адъювантов
131. Применимы требования, изложенные в пунктах 67 - 71 настоящей главы.
6.11. Исследование стабильности и срок годности (срок хранения)
132. Применимы требования, изложенные в пунктах 114 и 115 настоящей главы.
133. Необходимо разработать протокол проведения исследований стабильности вакцины готовности к пандемии. Необходимо описать и обосновать процедуру, согласно которой для вакцины и промежуточных продуктов производства определяется срок годности (срок (хранения) и дата истечения срока годности (срока хранения). Продлевать срок годности (срок хранения) необходимо на основании данных исследования стабильности в реальном времени.
Исследование стабильности после вскрытия упаковки
134. Применимы требования, изложенные в пункте 116 настоящей главы.
7. Требования к качеству при подаче заявления о внесении изменений в состав пандемических гриппозных вакцин (изменение пандемического штамма) во время пандемии
135. После официального признания пандемии (объявления ВОЗ в установленном порядке пандемической ситуации или объявления соответствующими уполномоченными органами государств-членов эпидемии, вызванной пандемическим типом вируса гриппа) заявитель вправе представить в уполномоченный орган (экспертную организацию) референтного государства заявление о внесении изменений в состав пандемических гриппозных вакцин (об изменении пандемического штамма) в целях включения объявленного пандемического штамма в пандемическую вакцину (обновление пандемического штамма) в соответствии с приложением № 24 к Правилам регистрации и экспертизы.
7.1. Штаммы-кандидаты для производства пандемической вакцины
136. В случае, если документация, описанная в пунктах 15 - 22 и 92 - 98 настоящей главы недоступна к моменту подачи заявления о внесении вызвавшего пандемию штамма в состав пандемической вакцины в полном объеме ее необходимо представить в кратчайшие возможные сроки после получения недостающих данных. Необходимо представить оценку рисков для вакцины, учитывая применяемую стратегию описания процесса производства и его контроля.
7.2. Качество и оценка качества вирусного посевного материала
137. Применимы требования, изложенные в пунктах 23 - 34 и пунктах 99 - 101 настоящей главы.
138. До тех пор, пока один из сотрудничающих центров ВОЗ не представит специфичные антисыворотки, для подтверждения подлинности посевного вируса необходимо разработать и использовать альтернативные методы испытаний (например, ПЦР, секвенирование, по крайней мере, участка генома, ответственного за синтез НА). После появления реактивов для подтверждения подлинности посевного вируса испытания проводятся с использованием реакции торможения гемагглютинации.
139. Необходимо обосновать любые изменения посевного вируса для вакцины, внесенные в процессе производства пандемической вакцины (например, переход на более продуктивный реассортантный вирус или дополнительный пассаж от того же производственного штамма). Краткий обзор сопроводительных данных представлен в таблице.
Таблица
Данные, которые необходимо представить при внесении изменений в вирусный посевной материал при производстве пандемической вакцины
| Этап производства | Требования к представляемым данным |
|---|---|
| Посевной вирус | подтверждение соответствия спецификациям, (например, испытание на подлинность (методами РТГА, РИНА)). В случае если используются штаммы, полученные с помощью методов обратной генетики, необходимо подтвердить, что у гена НА посевных материалов после дополнительного пассажа сохраняется та же самая генетическая последовательность, что и у вируса главного посевного материала и штамма-кандидата для производства вакцины, представленного сотрудничающим центром или аккредитованной контрольной лабораторией ВОЗ или другой официальной лабораторией, чтобы гарантировать, что безопасность и иммуногенность остались на неизменном уровне |
| Вирусный сбор | испытания по оценке выхода антигена НА |
| Активная фармацевтическая субстанция (моновалентный сбор) | валидация процесса всех важнейших этапов производства, признанных специфичными для определенного штамма. Данные об определении показателей спецификации. Результаты испытаний серий. Первые 3 серии моновалентных нерасфасованных продуктов, полученных из нового рабочего посевного материала необходимо проверить на наличие и тип антигена НА и NA. Данные о стабильности для обоснования заявленного срока годности (срока хранения) (необходимо представить данные плана-графика испытаний, если полных данных еще нет) |
| Готовая вакцина | результаты испытаний серии. Данные о стабильности для обоснования заявленного срока годности (срока хранения) (необходимо представить данные плана-графика испытаний, если полных данных еще нет) |
140. Любые различия, наблюдаемые при исследованиях готовой вакцины (например, в выходе продукта, содержании остаточных веществ (например, дезоксихолата натрия)), необходимо оценивать критически. Их необходимо обосновать (с точки зрения влияния на безопасность и иммуногенность вакцины) исходя из опыта, полученного ранее, при изготовлении вакцин для профилактики гриппа. Необходимо инициировать проведение дополнительных исследований влияния результатов испытаний для вирусных посевных материалов разных штаммов (например, исследований, направленных на оценку процесса инактивации и полноты инактивации).
7.3. Разработка производственного процесса
141. Применимы указания, изложенные в пунктах 103 и 104 настоящей главы.
142. Необходимо представить подробное описание, обоснование и данные о валидации изменений, которые вносятся в процесс производства (например, предназначенные для увеличения выхода продукции, масштабирования процесса производства).
143. Если данные по иммуногенности для человека еще не получены, изменение штамма-кандидата для производства вакцины необходимо обосновать соответствующими данными об иммуногенности, полученными при использовании лабораторных животных, по крайней мере, для одной серии.
7.4. Валидация производственного процесса
144. Применимы требования, изложенные в пунктах 43 - 49 настоящей главы.
7.5. Описание характеристик вакцины
145. Применимы требования, изложенные в пунктах 50 - 58 настоящей главы.
146. Поскольку полная сопоставимость вакцины не будет установлена до и после замены пандемического штамма, необходимо проводить сравнение важнейших показателей ее качества. Любые различия необходимо рассматривать с точки зрения безопасности и иммуногенности вакцины.
7.6. Стандартизация вакцины
147. При соответствующем обосновании и подтверждении результатами клинических исследований, вакцина на основе потенциально пандемического штамма может содержать количество гемагглютинина, отличное от 15 мкг для каждого из штаммов.
148. До появления реактивов для одиночной радиальной иммунодиффузии, для определения специфической активности вакцины с потенциально пандемическим штаммом необходимо пользоваться альтернативными методами испытаний. После того, как реактивы станут доступны, для определения специфической активности необходимо использовать метод одиночной радиальной иммунодиффузии.
7.7. Оценка адъювантов
149. Применимы требования, изложенные в пунктах 67 - 71 настоящей главы.
7.8. Изучение стабильности и срок годности (срок хранения)
150. Соответствующие исследования стабильности активной фармацевтической субстанции и вакцины проводятся в соответствии с протоколом, разработанным непосредственно для вакцины, содержащей потенциально пандемический штамм. Необходимо определить сроки и условия хранения для пандемической вакцины, содержащей штамм, объявленный причиной пандемии. На момент подачи заявления о внесении пандемического штамма в состав пандемической вакцины, данные об исследованиях стабильности серий промышленного масштаба в реальном времени могут быть неполными. В качестве данных, подтверждающих стабильность, используются результаты испытаний опытно-промышленных серий, если такие материалы являются репрезентативными для полномасштабного процесса производства. Заявленный срок годности (срок хранения) подтверждаются путем сравнения доступных данных о стабильности, полученных в испытаниях естественного, ускоренного хранения и (или) стресс-исследования, которые проводятся для вакцин до и после обновления пандемического штамма.
151. О результатах, выходящих за рамки спецификации, или об отклоняющихся данных для любого из аспектов качества необходимо сообщать уполномоченным органам (экспертным организациям) государств-членов.
152. Продлевать срок годности (срок хранения) необходимо на основании данных изучения стабильности в реальном времени.
Исследование стабильности после вскрытия упаковки
153. Исследование стабильности после вскрытия упаковки должны проводиться в соответствии с протоколом, согласованным до обновления пандемического штамма (например, для вакцины, содержащей потенциально пандемический штамм), чтобы подтвердить заявленный срок применения пандемической вакцины после смешивания препаратов антигена и системы адъюванта (если применимо).
8. Требования к качеству при подаче заявления о регистрации пандемической вакцины во время пандемии (экстренная процедура)
154. После объявления ВОЗ пандемической ситуации или объявления соответствующими уполномоченными органами государств-членов эпидемии, вызванной пандемическим типом вируса, регистрация новой пандемической вакцины осуществляется в экстренном порядке в соответствии с требованиями, изложенными в приложении № 24 к Правилам регистрации и экспертизы. Также применимы требования, изложенные в пунктах 119 - 121 настоящей главы в отношении уже объявленного пандемическим штамма.
9. Живые аттенуированные вакцины для профилактики гриппа
155. Живые аттенуированные вакцины для профилактики гриппа получают путем репродукции аттенуированных реассортантных вакцинных вирусов гриппа в развивающихся куриных эмбрионах. Для производства живых аттенуированных вакцин для профилактики гриппа допускается использовать реассортантные вакцинные штаммы, имеющие генетические маркеры аттенуации: холодоадаптированный или температурочувствительный фенотип. В отличие от инактивированных вакцин при производстве живых вакцин процессы очистки не применяются. Производство живых аттенуированных вакцин требует ужесточения оценки и контроля исходного сырья, субстрата для культивирования вируса, аттенуированного родительского штамма, штамма-донора гемагглютинина и нейраминидазы. В целях исключения любого потенциального источника контаминации исходное сырье, используемое при производстве вакцин для медицинского применения должно быть сертифицированным и удовлетворять нормативным требованиям. В связи с ежегодным изменением штаммового состава, вакцины для профилактики гриппа постоянно претерпевают изменения, что требует проведения тестов по оценке пригодности исходного сырья.
10. Требования к качеству при подаче заявления о регистрации живых гриппозных вакцин для профилактики сезонного гриппа
10.1. Аттенуированный родительский штамм (донор аттенуации) и его разработка
156. Сведения об аттенуированном родительском штамме: разработке, характеристике, вирусном посевном материале донора аттенуации необходимо представить в разделе 3.2.S.2.3 модуля 3 регистрационного досье.
Разработка аттенуированного родительского штамма
157. Необходимо представить детальную, документально подтвержденную историю и (или) качество всех биологических агентов, включая используемые штаммы вирусов, и субстраты их культивирования, культуры клеток (если применимо), а также валидационные протоколы всех этапов технологии приготовления и полный перечень всех исходных материалов, использованных при разработке аттенуированного родительского штамма (донора аттенуации).
Характеристика аттенуированного родительского штамма
158. В регистрационном досье должны быть представлены фенотипические и генотипические свойства аттенуированного родительского штамма.
159. Характеристика фенотипических свойств включает в себя изучение маркеров аттенуации (в исследованиях in vivo), ключевые фенотипические маркеры: холодоадаптированный (са) фенотип или температурочувствительный (ts) фенотип в исследованиях in vitro, позволяющих оценить возможность реверсии специфических аттенуирующих мутаций в «дикий» тип или реассортации донора аттенуации с «диким» вирусом.
160. Должны быть представлены материалы, подтверждающие отсутствие любого рода нейровирулентных свойств аттенуированного родительского штамма. Исследования нейровирулентности должны быть проведены с использованием животных, обладающих наиболее высокой восприимчивостью к заражению вирулентными штаммами вируса гриппа. Необходимо оценить не только потенциальную прямую нейровирулентность, опосредованную собственно аттенуированным родительским штаммом, но и косвенную, обусловленную вторичными инфекциями. Возможность использования мелких лабораторных животных при изучении нейровирулентности должна быть обоснована.
161. Генетическая характеристика аттенуированного родительского штамма включает в себя:
определение нуклеотидного состава всего вирусного генома с использованием секвенирования;
отчеты анализов молекулярных основ аттенуированного фенотипа;
оценку генетической стабильности аттенуированного родительского штамма путем сравнения нуклеотидной последовательности вирусного генома на разных уровнях пассажа.
162. Особое внимание заявителем должно быть уделено доказательству отсутствия посторонних агентов в вакцине. Такие исследования проводятся согласно требованиям к оценке вероятности присутствия посторонних агентов в вирусных посевных материалах, клеточных субстратах, а также требованиям к испытаниям на стерильность и на микоплазмы.
Вирусный посевной материал аттенуированного родительского штамма
163. Производство живой аттенуированной вакцины для профилактики гриппа основывается на системе посевных вирусов (seed-lot).
164. Посевной материал аттенуированного родительского штамма (донора аттенуации), используемый для получения реассортантных вакцинных штаммов для соответствующего вида живой гриппозной вакцины, культивируют в развивающихся куриных эмбрионах категории SPF. Метод получения должен быть детально описан, методы контроля инфекционной активности, генетической стабильности и стерильности посевных вирусов в заявленных условиях хранения должны быть валидированы.
165. Кроме того, в случае использования технологии методов обратной генетики, также необходимо иметь банки кДНК-клонов для шести фрагментов РНК, полученных от родительского штамма (М, NS, NP, РА, РВ1 и РВ2) в целях производства реассортантного вируса.
166. Должна быть разработана и введена программа по изучению стабильности посевных вирусов.
167. Для посевного материала аттенуированного родительского штамма требуется проведение контроля на стерильность и отсутствие микоплазм в соответствии с требованиями Фармакопеи Союза, а при отсутствии в ней - с требованиями фармакопей государств-членов. Клеточный субстрат, используемый для разработки производственного штамма-кандидата для вакцины, должен соответствовать требованиям Фармакопеи Союза, а при отсутствии в ней - требованиям фармакопей государств-членов.
168. В случае согласования с уполномоченным органом (экспертной организацией) государства-члена данные тесты проводятся на уровне рабочего посевного материала реассортантного вакцинного штамма.
10.2. Вирусы гриппа «дикого» типа как доноры НА и NA и их разработка
169. Сведения о вирусе гриппа дикого типа (доноре НА и NA): выделении и истории пассажа, вирусном посевном материале штамма-донора НА и NA необходимо представить в разделе 3.2.S.2.3 модуля 3 регистрационного досье.
Выделение и история пассажа штамма-донора НА и NA дикого типа
170. Поверхностные антигены НА и NA вируса дикого типа должны соответствовать антигенной структуре штамма вируса гриппа, рекомендованного ВОЗ на текущий эпидемический сезон. Происхождение и история пассажей штамма должны быть документированы надлежащим образом.
171. Необходимо обеспечить, чтобы антигенное подобие штамма-донора НА и NA вируса гриппа дикого типа и штамма, рекомендованного ВОЗ было документально подтверждено сотрудничающим центром ВОЗ (с помощью перекрестной реакции торможения гемагглютинации).
Вирусный посевной материал штамма-донора НА и NA
172. Посевной материал штамма-донора НА и NA должен быть получен от штамма, который был изолирован в развивающихся куриных эмбрионах или в контролируемой и сертифицированной клеточной линии. Куриные эмбрионы должны быть получены от поголовья кур категории SPF.
Качество вирусного посевного материала штамма-донора НА и NA
173. Производителю необходимо провести испытание подлинности гемагглютинина и нейраминидазы.
174. Тесты, проводимые в соответствии с Фармакопеей Союза, а при отсутствии в ней - в соответствии с фармакопеями государств-членов должны подтвердить отсутствие посторонних агентов в вирусном посевном материале штаммов-доноров НА и NA. Должны быть проведены специфические анализы на отсутствие человеческих респираторных патогенов, способных размножаться в развивающихся куриных эмбрионах. В дополнение к этим тестам разрабатываются тесты на основе мультиплексной ПЦР с целью выявления посторонних респираторных вирусов, которые могут реплицироваться в развивающихся куриных эмбрионах, используемых для получения серий посевных вирусов. Методы контроля должны быть валидированы.
175. Описанные в пунктах 173 и 174 настоящей главы тесты должны быть выполнены на заключительном этапе, когда штамм-донор НА и NA дикого типа используется для реассортации совместно с живым аттенуированным родительским штаммом (донором аттенуации).
В случае согласования с уполномоченным органом (экспертной организацией) референтного государства данные тесты проводятся на уровне реассортантного рабочего посевного вируса.
176. Так как элиминация или инактивация микробных контаминантов невозможна ни на одной из производственных стадий приготовления живой аттенуированной вакцины, их наличие недопустимо в посевном материале живого аттенуированного родительского штамма и штамма-донора НА и NA дикого типа.
10.3. Разработка реассортантного вакцинного штамма для производства живых гриппозных вакцин
177. Сведения о разработке, культивировании и получении посевного материала, главном и рабочем посевном материале реассортантного вакцинного штамма необходимо представить в разделе 3.2.S.2.3 модуля 3 регистрационного досье.
178. Аттенуированные реассортантные вакцинные штаммы получают с использованием метода классической реассортации между донором аттенуации и штаммом-донором НА и NA дикого типа или метода обратной генетики. Биологические реагенты, такие как антисыворотка или ферменты, используемые при получении реассортантного вакцинного штамма, должны быть проверены на отсутствие посторонних инфекционных агентов и их качество должно быть подтверждено соответствующими результатами анализа.
179. Наличие поверхностных антигенов - НА и NA от эпидемически актуального вируса гриппа дикого типа (донора НА и NA) в реассортантном вакцинном штамме должно быть подтверждено с использованием специфической антисыворотки и эталонных реагентов, имеющих соответствующие сертификаты, либо другим методом.
Культивирование и получение посевного материала реассортантного вакцинного штамма
180. Охарактеризованные клоны аттенуированного реассортантного вакцинного штамма культивируют в развивающихся куриных эмбрионах, свободных от специфической патогенной микрофлоры (SPF). Производство вакцины основывается на системе посевных вирусов. Из реассортантного вакцинного штамма готовится главный посевной вирус (главный посевной реассортантный вакцинный штамм). Из главного посевного вируса готовят рабочий посевной вирус (рабочий посевной реассортантный вакцинный штамм).
Характеристика главного посевного и рабочего посевного материала реассортантного вакцинного штамма
181. Главный посевной материал и рабочий посевной материал должны быть охарактеризованы в соответствии с требованиями указанными в пунктах 156 - 162 настоящей главы. Характеристика главного и рабочего посевных материалов должна содержать информацию о наличии генетических маркеров аттенуации, а также поверхностных антигенов - НА и NA от эпидемически актуального вируса гриппа «дикого» типа.
182. Сотрудничающий центр ВОЗ должен проверить и зафиксировать документально соответствие антигенной специфичности главного вирусного посевного материала штамму, рекомендованному ВОЗ (с использованием метода перекрестной реакции торможения гемагглютинации).
183. Каждый новый рабочий посевной материал должен оцениваться на генетическую стабильность методом секвенирования. Исследования генетической стабильности реассортантных вакцинных штаммов включают в себя изучение сохранения определенных фенотипических характеристик и генетической структуры аттенуированного родительского штамма на протяжении большего числа пассажей культивирования посевного материала (минимум после пятикратного пассирования).
184. Должно быть подтверждено отсутствие нейровирулентности реассортантного вакцинного штамма. В случае если это испытание не проводится или используются другие альтернативные испытания необходимо представить обоснование.
10.4. Субстрат для культивирования реассортантного вакцинного штамма для производства вакцины
185. Посевной материал вируса гриппа, используемый в производстве вакцин культивируют в развивающихся куриных эмбрионах или в подходящих культурах клеток, таких как фибробласты куриных эмбрионов или клетки почки цыпленка, полученных из стад категории SPF, или в диплоидных или перевиваемых линиях клеток, качество которых должно соответствовать требованиям общей фармакопейной статьи на вакцины для медицинского применения Фармакопеи Союза, а при отсутствии в ней - требованиям общих фармакопейных статей (монографий) на вакцины для медицинского применения фармакопей государств-членов.
186. Куриные эмбрионы, которые предстоит использовать для производства вакцины, должны быть взяты от здорового поголовья кур категории SPF или должны быть получены из птицехозяйств, благополучных по возбудителям, патогенным для человека (качество поставляемых эмбрионов должно быть подтверждено ветеринарными свидетельствами и входным контролем на отсутствие возбудителей, патогенных для человека (аденовирус, микоплазма, лейкоз птиц) или наличием документа производителя куриных эмбрионов, подтверждающего отсутствие возбудителей, патогенных для человека).
10.5. Производство вакцины
187. Сведения, касающиеся производства вакцины, необходимо представить в разделах 3.2.S.2 и 3.2.P.3 модуля 3 регистрационного досье.
188. Все этапы производства вакцины должны осуществляться с соблюдением требований Правил производственной практики. Обязательным требованием является соблюдение асептических условий производства, исключающих возможность контаминации промежуточных продуктов и полуфабрикатов посторонними инфекционными агентами.
189. При проведении испытаний на посторонние агенты развивающихся куриных эмбрионов, используемых в производственном цикле, параллельно инкубируют контрольные развивающиеся куриные эмбрионы без инокулята и проверяют их на отсутствие посторонних агентов.
190. В качестве альтернативы допускается проверять отдельные сборы на посторонние агенты на подходящих клеточных субстратах в присутствии антител, нейтрализующих гемагглютинин вируса гриппа.
Куриные эмбрионы, используемые в качестве субстрата для производства вакцины
191. Развивающиеся куриные эмбрионы, используемые в качестве субстрата для получения вакцины, должны быть взяты из поголовья птиц, строго контролируемых на соответствие категории SPF или должны быть получены из птицехозяйств, благополучных по возбудителям, патогенным для человека (качество поставляемых эмбрионов должно быть подтверждено ветеринарными свидетельствами). В приготовлении живой аттенуированной вакцины для профилактики гриппа должно быть использовано поголовье прошедшее отбор указанным способом.
Вирусный сбор
192. Использование тиомерсала в живой аттенуированной вакцине для профилактики гриппа, как и для любой живой вирусной вакцины, не допускается. В последующих производственных циклах должен использоваться вирусный сбор, в отношении которого подтверждены:
антигенная специфичность (то есть, определена принадлежность его НА вирусу «дикого» типа (штамму донору НА и NA));
отсутствие нейровирулентности.
Тесты на стерильность и отсутствие микоплазм, в случае согласования с уполномоченным органом референтного государства, проводятся на следующем этапе производственного процесса. В спецификацию на данном этапе должна быть включена оценка инфекционной активности, то есть определение эмбриональной инфекционной дозы, выражающейся в ЭИД50 в 1 мл вируссодержащей аллантоисной жидкости или оценка меры инфекционной активности количественным способом в специальных единицах формирования фокуса на миллилитр (FFU/мл) (выполненная методом флуоресцирующих антител).
Моновалентный нерасфасованный продукт
193. Испытания для моновалентного нерасфасованного продукта включают в себя испытания на сохранение генетических маркеров и подтверждение фенотипа живого аттенуированного вируса. Характеристика фенотипических свойств включает изучение маркеров аттенуации (например, маркер холодоадаптированности (или холодоадаптированный (са) фенотип) или маркер температурочувствительности (температурочувствительный (ts) фенотип)) в исследованиях in vivo и (или) in vitro, позволяющих оценить возможность реверсии специфических аттенуирующих мутаций в дикий тип или реассортации донора аттенуации с диким вирусом.
194. Необходимо провести испытания подлинности гемагглютинина и нейраминидазы.
Тривалентный нерасфасованный продукт и готовая вакцина
195. Тривалентный нерасфасованный продукт в соответствии со спецификацией должен быть изучен на специфическую активность.
196. Такие показатели качества, как специфическая активность, содержание овальбумина и бактериальных эндотоксинов должны входить в спецификацию на готовую вакцину (раздел 3.2.Р.5.1 модуля 3 регистрационного досье).
197. Термостабильность готовой вакцины необходимо подтверждать соответствующим образом как в режиме реального времени, так и в условиях повышенной температуры. Необходимо определить и должным образом обосновать показатели спецификации в конце срока годности (срока хранения).
10.6. Валидация производственного процесса
198. Необходимо представить данные по валидации производственного процесса с целью подтверждения того, что критические стадии производства при установленных параметрах, способны эффективно и воспроизводимо обеспечить получение готовой вакцины, отвечающей требованиям, изложенным в спецификации.
199. Описание, документацию и результаты исследований по валидации производственного процесса, критических точек производства или методов количественного определения, используемых в производственном процессе, необходимо представить в разделах 3.2.S.2.5 и 3.2.Р.3.5 модуля 3 регистрационного досье.
10.7. Описание характеристик вакцины
200. Определение характеристик вакцины необходимо для установления соответствующих спецификаций и обеспечения научно обоснованной оценки сопоставимости после внесения изменений в вакцину или процесс ее производства.
201. Биологические, иммунологические и физико-химические свойства антигена НА должны быть изучены с использованием широкого спектра современных аналитических методов. Например, должна быть оценена агрегация частиц, фенотип (генотип), морфология вируса, специфическая активность. Необходимо провести испытание подлинности и количественного определения технологических примесей (например, овальбумина (остаточного белка клеток-продуцентов)) и появившихся на более поздних этапах процесса производства примесей. Полученные данные необходимо использовать для уточнения показателей и норм спецификации для выпуска готовой вакцины.
10.8. Форма выпуска
202. В случае необходимости разработки определенной формы выпуска для особой целевой популяции разработка такой формы выпуска должна быть обоснована и подтверждена соответствующими данными о качестве, которые включают в себя информацию о совместимости, валидации производственного процесса и стабильности.
10.9. Стандартизация вакцины
203. Спецификация на готовую вакцину должна содержать данные о специфической активности (ЭИД50 или ЦПД50 (цитопатогенное действие)). Специфическую активность определяют для каждого серотипа, входящего в состав вакцины, с использованием референс-реагентов (для моновалентного нерасфасованного продукта - раздел 3.2.S.4.1, для готового продукта (вакцины) - раздел 3.2.Р.5.1 модуля 3 регистрационного досье).
10.10. Изучение стабильности и срок годности (срок хранения)
204. Должны быть представлены результаты испытания стабильности естественного, ускоренного хранения и (или) стресс- исследования, для моновалентных нерасфасованных продуктов (разделы 3.2.S.7.1 - 3.2.S.7.3 модуля 3 регистрационного досье) и готовой вакцины (разделы 3.2.Р.8.1 - 3.2.Р.8.3 модуля 3 регистрационного досье). Должен быть разработан протокол оценки стабильности вакцины. Сроки и условия хранения должны быть обоснованы. Любое продление срока годности (срока хранения) должно основываться на данных исследования стабильности в режиме реального времени.
11. Требования к качеству при подаче заявления об изменении штаммового состава живых аттенуированных вакцин для профилактики сезонного гриппа
11.1. Вирусы гриппа «дикого» типа как доноры НА и NA и разработка реассортантного вакцинного штамма
205. История производства посевного материала, должна включать в себя следующие документы и данные:
описание процедуры получения посевного материала (начиная с главного донора-аттенуации и штамма рекомендованного ВОЗ);
историю пассажа;
генетическую последовательность посевного материала;
описание фенотипических характеристик, включающее в себя результаты изучения маркеров аттенуации (в исследованиях in vivo), ключевых фенотипических маркеров: холодоадаптированный (са) фенотип или температурочувствительный (ts) фенотип (в исследованиях in vitro, позволяющих оценить возможность реверсии специфических аттенуирующих мутаций в «дикий» тип или реассортации донора аттенуации с «диким» вирусом);
генетическую стабильность посевного материала, включая актуальные генотипические и фенотипические маркеры (секвенирование полного генома);
антигенное подобие штамма-донора дикого типа и главного посевного материала штамму, рекомендованному ВОЗ (проводится с помощью перекрестной реакции торможения гемагглютинации), которое документально подтверждено сотрудничающим центром ВОЗ;
протоколы аналитических испытаний (включая испытания на отсутствие посторонних агентов). В случае если посевной вирус проверяется на посторонние агенты методом ПЦР и при обсуждении вопроса с уполномоченным органом (экспертной организацией) референтного государства была согласована необходимость проведения дополнительных ПЦР-испытаний посевного материала, эти данные необходимо включить в регистрационное досье вакцин для профилактики гриппа;
испытания нейровирулентности. При ежегодном изменении (обновлении) штаммового состава (то есть для антигенно дрейфовых вариантов вируса гриппа) испытания нейровирулентности допускается не проводить. Такое испытание проводится в случае, если в вакцину включается новый подтип гемагглютинина вируса гриппа А (то есть не подтипа Н1 или НЗ) или новый вирус гриппа В, отличающийся генетически от циркулирующих на данный момент линий, или в случае возникновения особых опасений о безопасности.
11.2. Разработка производственного процесса
206. Любые изменения, связанные с оптимизацией производственных процессов, обусловленные специфическими характеристиками штамма необходимо обосновать, соответствующие сведения необходимо включить в раздел 3.2.S.2 модуля 3 регистрационного досье.
207. Необходимо представить в разделе 3.2.Р.3.2 модуля 3 регистрационного досье подробный состав на серию (производственную рецептуру) в связи с изменением (обновлением) штаммового состава и описание вакцины и ее состав в разделе 3.2.Р.1 модуля 3 регистрационного досье.
208. Если уполномоченным органом (экспертной организацией) государства-члена было вынесено заключение о необходимости проведения клинического исследования в связи с изменением штаммового состава, то необходимо представить сертификат соответствия серий, используемых в клинических исследованиях. Сертификат соответствия серий представляется в комплекте документов по качеству либо в комплекте документов по клиническим исследованиям. Соответствующие сведения необходимо включить в раздел 3.2.Р.2.2.1 модуля 3 регистрационного досье.
11.3. Валидация производственного процесса и (или) его оценка
209. При изменении (обновлении) штаммового состава сезонных вакцин для профилактики гриппа необходимо представить результаты исследований по валидации для критических точек производства в разделе 3.2.S.2.5 модуля 3 регистрационного досье.
210. Результаты анализа серии первых 3 моновалентных нефасованных продуктов, полученных из каждого нового посевного материала, должны быть представлены в разделе 3.2.S.4.4 модуля 3 регистрационного досье.
211. Должны быть представлены результаты анализа серии вакцины, включая оценку термостабильности.
11.4. Стандартизация вакцины
212. Необходимо представить результаты валидации аналитических методик, на изменение которых может повлиять замена одного или нескольких штаммов (например, валидация метода определения специфической активности). Данные о валидации необходимо представить в разделе 3.2.S.4.3 для моновалентных нефасованных продуктов, а также трехвалентных нерасфасованных продуктов или вакцины в разделе 3.2.Р.5.3 модуля 3 регистрационного досье.
213. Необходимо представить в форме таблиц копии утвержденных спецификаций для моновалентных нефасованных продуктов в разделе 3.2.S.4.1 модуля 3 регистрационного досье и для вакцины в разделе 3.2.Р.5.1 модуля 3 регистрационного досье, а также обзор аналитических методик в разделе 3.2.S.4.2 модуля 3 регистрационного досье.
11.5. Изучение стабильности и срок годности (срок хранения)
214. Необходимо представить результаты испытаний стабильности моновалентных нерасфасованных продуктов если предполагается их использование более 1 года. Соответствующие сведения включают в раздел 3.2.S.7 модуля 3 регистрационного досье.
215. Для лекарственного препарата (готовой вакцины), необходимо представлять в разделе 3.2.Р.8 модуля 3 регистрационного досье данные о стабильности как за предыдущий сезон, так и обязательство продолжать исследования стабильности нескольких серий лекарственного препарата по программе, описанной в протоколе пострегистрационных исследований стабильности.
216. Результаты испытаний стабильности методом «ускоренного старения» используются для того, чтобы показать, что новые штаммы, вероятно, будут иметь те же характеристики стабильности, что и штаммы, которые используются в исследованиях стабильности, на которых основан срок годности (срок хранения).
ПРИЛОЖЕНИЕ № 1
к главе 28 Правил проведения
исследований биологических
лекарственных средств
Евразийского экономического союза
УКАЗАНИЯ
по качеству штаммов-кандидатов для производства вакцин для профилактики гриппа, полученных в культурах клеток
1. Общие положения
1. В настоящем документе изложены указания, касающиеся требований к качеству культур клеток, используемых для выделения штаммов-кандидатов для производства вакцин для профилактики гриппа; условиям, при которых выделяются вирусы, последующего пассирования вирусов до того момента как производителем будет подготовлен главный посевной материал.
2. Многие производители инактивированных вакцин для профилактики гриппа разрабатывают собственные процессы культивирования вирусов в подходящих культурах клеток. Для получения посевного материала выбираются рекомендованные ВОЗ штаммы-кандидаты, полученные с использованием развивающихся куриных эмбрионов: изолят дикого типа, выделенный в развивающихся куриных эмбрионах или высокоурожайный реассортант, особенно для вируса гриппа типа А. Реассортанты и штаммы-кандидаты дикого типа получают в одной из специализированных лабораторий ВОЗ, сотрудничающих центрах ВОЗ по гриппу и других сертифицированных лабораториях, специализирующихся на разработке штаммов-кандидатов.
3. Наибольшее предпочтение при производстве «культуральных» вакцин отдается штаммам-кандидатам, выделенным в культуре клеток вместо штаммов-кандидатов, выделенных в развивающихся куриных эмбрионах, поскольку вирус гриппа человека, адаптированный для репродукции в развивающихся куриных эмбрионах подвергается фенотипическим и генотипическим изменениям. Вирусы гриппа человека, выделенные и в последующем пассируемые в этих же культурах клеток млекопитающих, по сравнению с репродукцией в развивающихся куриных эмбрионах менее подвержены таким изменениям, и, как правило, гемагглютинин вируса, который выделен и размножен в культуре клеток, генетически и фенотипически ближе к вирусу, обнаруживаемому в клинических изолятах, в отличие от адаптированных к развивающимся куриным эмбрионам вариантов, в которых были идентифицированы специфические замены аминокислот гемагглютинина.
4. Производители культуральных вакцин для профилактики гриппа как правило не используют адаптированные к репродукции в развивающихся куриных эмбрионах вирусы, которые с точки зрения антигенов отдалены от вирусов гриппа дикого типа. Некоторые сотрудничающие центры ВОЗ разрабатывают штаммы-кандидаты в линиях клеток, аттестованных для получения этих штаммов-кандидатов для производства вакцин.
5. Основная проблема получения штаммов-кандидатов для производства вакцин в аттестованных линиях клеток - возможность контаминации посторонними агентами, которые могут потенциально присутствовать в источнике культуре клеток, материалах используемых при выделении и культивировании производственного штамма-кандидата. В настоящем документе описаны требования к:
качеству культур клеток, которые применяются при выделении вируса;
условиям, при которых изолируют вирусы;
последующему пассированию этих вирусов до получения главного посевного материала производителем.
6. Настоящий документ распространяется на штаммы-кандидаты, выделенные в культуре клеток для производства вакцин с использованием в качестве субстрата для репродукции вируса культуры клеток или развивающихся куриных эмбрионов.
7. Производителям необходимо учитывать требования общей фармакопейной статьи на клеточные субстраты для производства вакцин для медицинского применения Фармакопеи Союза, а при отсутствии в ней - требования общих фармакопейных статей (монографий) фармакопей государств-членов.
8. Происхождение и история пассажей штаммов вируса должны быть одобрены уполномоченными органами (экспертной организацией) государств-членов.
2. Требования к клеточным субстратам, используемым для выделения вируса
9. В исследованиях вирусов гриппа и в разработке вакцин в качестве клеточных субстратов используют такие культуры клеток, как MDCK, Vero и первичных клеток, выделенных из кур. Если используется линия клеток, клетки необходимо получать из системы банка клеток.
10. Вирусная и микробиологическая безопасность клеточных линий животного происхождения должна отвечать требованиям общей фармакопейной статьи для производства вакцин для медицинского применения Фармакопеи Союза на клеточные субстраты, а при отсутствии в ней - требованиям общих фармакопейных статей (монографий) фармакопей государств-членов.
11. Должны быть известны происхождение клеточных субстратов, источник получения, в том числе характеристики питательной среды, которая использовалась для культивирования. Должны быть проведены испытания на подлинность и чистоту.
12. Проведение испытаний на туморогенность для линий клеток, для которых уже имеется соответствующая информация (например, MDCK, Vero, РегС.6 или первичные клетки, выделенные из птиц) не требуется.
13. Для выделения вируса гриппа используются культуры клеток, аттестованные для производства вакцин для медицинского применения, которые соответствуют требованиям Фармакопеи Союза, при отсутствии в ней - требованиям фармакопей государств-членов.
14. Производителю следует учитывать, что при использовании некоторых линий клеток (например, Vero), при культивировании и выделении из клинических изолятов вируса гриппа существует высокий риск одновременного культивирования и выделения сопутствующих вирусов человека.
3. Работа с клетками, выделение и культивирование вируса
15. Производителям необходимо тщательно подходить к вопросу описания состава и источника сред и всех манипуляций с клетками, в том числе при пассировании клеток, выделении и культивировании вируса. Необходимо использовать компоненты, неживотного происхождения. Если используются какие-либо материалы, полученные от человека или животных, в этих материалах не должно быть возбудителей инфекций. Бычью сыворотку, используемую для подготовки и культивирования культур клеток, необходимо подвергать воздействию рентгеновских лучей (X-ray). В отношении вирусной безопасности сырья биологического происхождения, необходимо представить соответствующие подтверждающие документы. Необходимо руководствоваться требованиями актов органа Союза по обеспечению безопасности при использовании бычьей сыворотки в процессе производства лекарственных препаратов и требованиям Фармакопеи Союза по минимизации риска передачи губчатой энцефалопатии животных.
16. Работу с вирусами необходимо выполнять в специально отведенном для этого боксе микробиологической безопасности в асептических условиях. Одновременно допускается работа только с одним вирусным изолятом.
17. Необходимо рассмотреть возможность более длительного промежутка очистки при работе с различными подтипами вируса гриппа. В лаборатории должна иметься специализированная система хранения клеток и штаммов-кандидатов для производства вакцины, описание процедуры работы (СОП). Информация о хранении банков клеток и системе используемых банков клеток должна быть документирована.
4. Обеспечение качества
18. Необходимо представить гарантии того, что все процедуры по культивированию клеток и вирусов выполняются полностью обученным (или проходящим обучение) персоналом в предназначенных для этого помещениях с использованием специального оборудования. Документация должна позволять полностью проследить применяемые технологические процедуры, работу оборудования, происхождение материалов и оценить подготовку персонала. Несмотря на то, что производители могут получать штаммы-кандидаты для производства вакцин из лабораторий ВОЗ, держатели регистрационных удостоверений несут ответственность за соответствие своего главного посевного материала требованиям качества для использования в процессе производства.
19. Если при производстве вакцины используется штамм-кандидат, полученный с использованием культур клеток, а в качестве субстрата для репродукции штамма-кандидата - развивающиеся куриные эмбрионы, то это не должно оказывать влияния на требования к качеству производимой в развивающихся куриных эмбрионах вакцины.
ПРИЛОЖЕНИЕ № 2
к главе 28 Правил проведения
исследований биологических
лекарственных средств
Евразийского экономического союза
ПРИМЕРЫ
описания разработки штамма-кандидата для производства вакцин для профилактики гриппа
I. Пример схемы описания разработки штамма-кандидата для производства вакцин для профилактики гриппа, полученного с помощью методов обратной генетики
Описание вируса: полученный с помощью метода обратной генетики реассортантный вирус с соотношением генов 2:6 от штаммов A/abc/123/04 и A/PR/8/34.
История пассажа: культура клеток Vero х1, развивающиеся куриные эмбрионы х1.
| Параметр | Стандартные операционные процедуры (применяемые методы) | Результаты (комментарии) |
|---|---|---|
| Репродукция штамма вируса гриппа A/abc/123/03 в развивающихся куриных эмбрионах | СОП xyz2, в условиях 4-го уровня биологической безопасности (BSL4) | вирус гриппа дикого типа, выращенный в развивающихся куриных эмбрионах, полученный из ... |
| Клонирование и генетическая модификация сегмента гемагглютинина штамма A/abc/123/04 | стандартные молекулярно-биологические методы | сегмент гемагглютинина штамма A/abc/123/04, клонированный с вырезанным полиосновным сайтом расщепления и с введенными стабилизирующими мутациями |
| Секвенирование клонированного гена гемагглютинина | секвенирование плазмидной ДНК | А/abc/123/04-подобный вирус без полиосновных аминокислот в сайте расщепления |
| Клонирование сегмента нейраминидазы штамма A/abc/123/04 | стандартные молекулярно-биологические методы | сегмент нейраминидазы штамма A/abc/123/04 клонирован без дальнейших модификаций |
| Секвенирование клонированного гена нейраминидазы | секвенирование плазмидной ДНК | А/abc/123/04-подобный вирус |
| Плазмиды PR8 | стандартные молекулярно-биологические методы | подготовлены ... или представлены ... |
| Плазмиды | СОП xyz2 | использованы плазмиды НАхх и NAzz, плюс 6 генов PR8 и 4 плазмиды-помощника |
| Клетки Vero | СОП xyz2 | клетки прошли валидацию для производства вакцин для медицинского применения |
| Обратная генетика | СОП xyz2 | реассортантный вирус выделенный из трансфецированных клеток Vero прошел 2 пассажа; титр гемагглютинина abc3 |
------------------------------
1 «х» - количество пассажей;
2 «xyz» - номер стандартной операционной процедуры;
3 «abc» - значение титра.
------------------------------
II. Пример спецификации штамма-кандидата, полученного с помощью метода обратной генетики
Описание вируса: полученный с помощью метода обратной генетики реассортантный вирус с соотношением генов 2:6 от штаммов A/abc/123/04 и A/PR/8/34.
История пассажа: клетки Vero х1, развивающиеся куриные эмбрионы х1.
| Параметр | Стандартная операционная процедура (метод контроля) | Референтные значения | Результат |
|---|---|---|---|
| Анализ антигенов вируса | СОП xyz2 | A/abc/123/04-подобный вирус | соответствуют спецификации |
| Титр вируса | СОП xyz2 | неприменима | гемагглютинирующий титр составляет ... |
| Инфекционная активность в развивающихся куриных эмбрионах | СОП xyz2 | неприменима | 10хЭИД50/мл |
| Последовательность гемагглютинина вируса | ПЦР с обратной транскрипцией (циклическое секвенирование) | А/abc/123/04-подобный вирус без полиосновных аминокислот в сайте расщепления | соответствует спецификации |
| Последовательность нейраминидазы вируса | ПЦР с обратной транскрипцией (циклическое секвенирование) | A/abc/123/04-подобный вирус | соответствует спецификации |
| Испытание патогенности для цыплят | СОП xyz2 | индекс внутривенной патогенности = 1,2 или ниже | результат составляет... соответствует спецификации |
| Испытание патогенности для хорьков | СОП xyz2 | титр вируса, выявляемый в органах дыхательной системы не должен быть выше, чем у родительских штаммов. Репликация вируса происходит только в дыхательных путях клинические симптомы (или их отсутствие) свидетельствуют об аттенуации вируса | соответствует спецификации |
| Испытание на куриных эмбрионах | СОП xyz2 | эмбрионы остаются живыми | соответствует спецификации |
| Стерильность | СОП xyz2 | отвечает требованиям | соответствует спецификации |
| Контаминация вируса плазмидной ДНК | ПЦД | неприменима | результат составляет ... |
| Материалы животного происхождения, использованные при получении вируса | Контроль материалов | соответствие Фармакопеи Союза по минимизации риска передачи губчатой энцефалопатии животных | соответствуют спецификация |
------------------------------
1 «х» - количество пассажей;
2 «xyz» - номер стандартной операционной процедуры.
------------------------------
Глава 29. Указания по проведению доклинических и клинических исследований вакцин для профилактики гриппа
1. Общие положения
1. Положения настоящей главы распространяются на процедуры регистрации вакцин для профилактики гриппа и определяют единый порядок проведения доклинических и клинических исследований при регистрации сезонных, пандемических и препандемических (зоонозных) вакцин для профилактики гриппа на таможенной территории Союза, а также при внесении изменений в регистрационное досье зарегистрированных сезонных, пандемических и препандемических (зоонозных) вакцин при изменении (обновлении) штаммового состава вакцин для профилактики гриппа.
2. Положения настоящей главы распространяются на следующие типы вакцин для профилактики гриппа:
живые аттенуированные вакцины для профилактики гриппа (ЖГВ);
инактивированные расщепленные, субъединичные и цельновирионные вакцины;
вакцины, содержащие адъюванты.
3. Положения настоящей главы также применимы к:
инактивированным вакцинам, содержащим альтернативные
вакцинные антигены (например, не содержащим цельные молекулы гемагглютинина);
вакцинам, содержащим рекомбинантные поверхностные антигены;
ДНК-вакцинам, экспрессирующим поверхностный антиген (антигены);
вакцинам на основе вирусоподобных частиц.
4. По вопросам регистрации в отношении всех новых вакцин, к которым положения настоящей главы не применяются в полной мере, заявители вправе обратиться за научной консультацией к уполномоченным органам (экспертным организациям) государств-членов.
5. К новым вакцинам относятся:
вакцины, которые сходны с зарегистрированной вакциной по типам антигенов и ожидаемому взаимодействию с иммунной системой (например, четырехвалентные инактивированные вакцины для профилактики гриппа, производимые аналогично трехвалентным инактивированным вакцинам);
вакцины, содержащие новую конструкцию или подход к технологии получения (например, консервативные белки или эпитопы белков вируса гриппа).
6. Настоящая глава дополняет требования Правил надлежащей клинической практики Евразийского экономического союза, Правил лабораторной практики и Правил практики фармаконадзора в части особенностей организации и проведения всех видов исследований вакцин для профилактики гриппа.
2. Определения
7. Для целей настоящей главы используются понятия, которые означают следующее:
«вакцина готовности к пандемии» - кандидатная вакцина (или технология приготовления вакцины) для профилактики гриппа, разрабатываемая в целях иммунизации населения в случае возникновения гриппа, вызванного пандемическими штаммами вируса гриппа;
«новая вакцина» - впервые регистрируемая вакцина для медицинского применения, имеющая иной антигенный состав, конструкцию или технологию получения по сравнению с уже зарегистрированными вакцинами на таможенной территории Союза;
«пандемическая вакцина» - вакцина для профилактики гриппа, предназначенная для иммунизации населения в случае возникновения гриппа, вызванного пандемическими штаммами вируса гриппа;
«препандемическая (зоонозная) вакцина» - вакцина для профилактики гриппа, содержащая новый штамм вируса гриппа животного происхождения с пандемическим потенциалом (зооноз - инфекционное заболевание, передающееся от животных человеку);
«сезонная вакцина» - вакцина для профилактики гриппа, вызываемого эпидемическими штаммами вируса гриппа, предназначенная для ежегодной иммунизации населения.
3. Требования к проведению доклинических исследований для регистрации сезонных вакцин всех типов для профилактики гриппа
3.1. Исследования первичной фармакодинамики
Оценка иммуногенности вакцин
8. Оценку иммуногенности вакцин для профилактики гриппа необходимо проводить с использованием мелких видов экспериментальных животных, наиболее чувствительных в отношении вакцин для профилактики гриппа (например, крыс, хомяков, морских свинок, мышей и хорьков).
9. Исследования иммуногенности вакцин для профилактики гриппа должны предусматривать:
оценку гуморального, а также клеточного иммунного ответа;
оценку соотношения «доза - эффект» путем исследования эффекта различных доз антигена при введении вакцины.
10. При разработке дизайна доклинических исследований необходимо принимать во внимание планируемый способ введения вакцины, поскольку он может повлиять на тип индуцируемого иммунного ответа. Иммунный ответ необходимо оценивать после введения каждой дозы вакцины. По результатам серологических исследований необходимо получить данные о кросс-нейтрализуюгцих антителах и кросс-реактивности пандемических, препандемических (зоонозных) и сезонных вакцин с адъювантом в отношении гетерологичных штаммов вирусов гриппа (оценка кросс-реактивности). Исследования иммуногенности на животных служат подтверждением воспроизводимости процесса производства, в частности, в ходе фазы валидации процесса производства разрабатываемой вакцины. Вместе с тем, учитывая принцип 3R (замена, улучшение и сокращение (replacement, refinement, reduction)) с целью минимизации количества привлекаемых к исследованию животных оценку иммуногенности целесообразно проводить также с использованием подходов in vitro или с учетом возможности получения соответствующих данных в клинических исследованиях.
Протективность (защитные свойства) вакцин
11. Оценку протективности, как наиболее надежный метод оценки специфической активности, необходимо проводить для гриппозных вакцин с новыми механизмами действия на релевантной модели животных. Эти исследования позволяют подтвердить защитную эффективность против штамма, входящего в состав разрабатываемой вакцины, предназначенной для клинических исследований.
12. Также исследования протективности необходимо проводить в тех случаях, когда соответствующие клинические данные при применении у человека отсутствуют (например, при разработке пандемических вакцин).
13. Наиболее адекватной моделью для исследования защитных свойств вакцин для профилактики гриппа (при условии, что исследуемый штамм вируса гриппа хорошо реплицируется и вызывает симптоматическую инфекцию) являются хорьки, поскольку патогенез заболевания, клиническая симптоматика, включая лихорадку, и механизмы формирования иммунитета схожи с таковыми у человека. Оценка протективности вакцин для профилактики гриппа на мышах нецелесообразна, поскольку хорьки обладают наиболее высокой восприимчивостью к заражению вирулентными штаммами вируса гриппа.
14. Вирус гриппа, используемый для заражения животных в ходе оценки протективной активности вакцин, должен соответствовать штамму дикого вируса, из которого получен вакцинный штамм. В исследованиях используют животных, не переносивших гриппозную инфекцию. В некоторых случаях хорьков необходимо праймировать штаммами другого серотипа вируса гриппа (например, если они являются «наивными» по определенным штаммам вируса гриппа или в случае использования слабоиммуногенных штаммов). В протокол исследования необходимо включить обоснование и информацию об исходном иммунном статусе хорьков.
15. Дизайн исследования может различаться в зависимости от типа исследуемой вакцины. Оценку протективности необходимо проводить при интраназальном способе заражения. При должном обосновании, также допустим интратрахеальный способ заражения. Для заражения животных предпочтительно использовать высокие инфекционные дозы вируса (~105 ИД50 или летальную дозу, если она известна). Основными конечными точками исследования протективности вакцин для профилактики гриппа являются:
неспецифические симптомы заболевания, такие как повышение температуры тела, изменение веса, аномалии в поведении животных, клинические проявления заболевания (например, чихание), повышение числа лейкоцитов, макроскопическая и гистологическая оценка органов, а также летальность;
инфекционные маркеры, такие как выделяемость вируса (с помощью назальных смывов, полученных в разное время), инфекционная активность вируса гриппа, кинетика репликации вируса (животных необходимо умерщвлять в разное время, извлекая образцы биологического материала как верхних, так и нижних дыхательных путей).
16. Как правило, не допускается предусматривать летальность в качестве единственной конечной точки протективности вакцины в исследованиях на хорьках, поскольку, с учетом принципов 3R (замена, улучшение и сокращение (replacement, refinement, reduction)), такие исследования не всегда предусматривают умерщвление животных.
17. Оценку кросс-протективности вакцин проводят путем заражения животных гетерологичным вирусом (вирусом отличным от вакцинного штамма). Такую оценку проводят для препандемических (зоонозных), пандемических и сезонных вакцин с адъювантом с целью подтверждения более широкой протективности.
Пассивная иммунизация
18. Исследования пассивной иммунизации животных позволяют оценить уровень защиты, индуцированный у «наивных» (неиммунизированных) животных после пассивного переноса антиген-специфических сывороток от иммунизированных животных или сывороток от вакцинированных людей. На основании полученных результатов делается вывод о том, может ли индуцированный вакциной гуморальный иммунный ответ обеспечить защиту от инфекции. Такие исследования особенно актуальны при оценке вакцин, предназначенных для невоспроизводимых (non-replicating) пандемических и зоонозных заболеваний, где целью является определение протективного титра антиген-специфических нейтрализующих антител.
3.2. Исследования фармакологической безопасности
19. Общие исследования фармакологической безопасности вакцин для профилактики гриппа, как правило, не требуются. Вместе с тем необходимо анализировать нежелательное действие на сердечно-сосудистую и дыхательную системы, а также на параметры центральной нервной системы, особенно при включении в состав вакцины адъюванта, или если эти органы являются мишенями для развития патогенеза вируса дикого типа (важно в отношении ЖГВ). Эти параметры необходимо включать в дизайн токсикологических исследований и исследований иммуногенности.
3.3. Фармакокинетические исследования
20. Исследования с целью определения концентрации антигенов в сыворотке не требуются. Специальные исследования проводятся с учетом типа вакцины, в отношении нового состава или адъювантов, либо при альтернативных способах введения (например, реакция в месте инъекции, исследования распределения и вирусовыделения в случае использования ЖГВ).
3.4. Токсикологические исследования
21. Токсикологические исследования необходимо проводить с использованием вакцины, содержащей тот же штамм что и разрабатываемая вакцина, предназначенная для применения в клинической практике.
22. При проведении доклинических исследований безопасности, уровни доз по содержанию антигена и вводимому объему должны быть эквивалентны дозе, предлагаемой для применения у человека. Вместе с тем необходимо представить научное обоснование адекватности выбранной дозы в отношении экспериментальных животных.
23. В отношении новых вакцин, процесс производства которых схож с процессом производства зарегистрированных вакцин, повторное проведение доклинических токсикологических исследований не требуется, в случае если эти исследования проведены в соответствии с Правилами надлежащей лабораторной практики, результаты представлены в полном объеме и имеют достаточную научную достоверность, а также представлено обоснование возможности экстраполяции данных на разрабатываемую вакцину.
24. Изучение токсичности при однократном введении предпочтительно проводить в рамках изучения токсичности при повторном (многократном) введении.
Исследования токсичности при повторном (многократном) введении
25. Токсикологические исследования разрабатываемой вакцины проводятся с использованием одного релевантного вида животных (например, крыс, хорьков, кроликов и т. д.). Дизайн исследования должен в наиболее полной мере отражать количество доз и частоту введения вакцины, запланированной при применении в клинических условиях. Интервалы дозирования могут быть короче (например, 2 - 3-недельными) с учетом специфики и различий в кинетике иммунного ответа, индуцируемого различными типами вакцин для профилактики гриппа.
26. В рамках исследования по изучению токсичности при повторном (многократном) введении, предусматривается включение конечных точек, необходимых для оценки иммунотоксических и аллергизирующих свойств вакцин (если не обосновано иное).
27. В случае использования адъювантов в составе вакцин требуется проведение оценки иммунотоксичности и реакций гиперчувствительности (в соответствии с главой 16 настоящих Правил).
Исследование репродуктивной токсичности
28. Изучение фертильной (эмбрио-фетальной, пренатальной) постнатальной токсичности должно включать как минимум 1 исследование с использованием одного вида животных. Дизайн исследования должен отражать планируемую схему клинического применения вакцины. Вакцинацию необходимо проводить перед спариванием животных и в ходе гестации.
Генотоксичность и канцерогенность
29. Исследования генотоксичности и канцерогенности вакцин для профилактики гриппа, как правило, проводить не требуется.
30. Отдельное внимание необходимо уделить адъювантам (в соответствии с главой 16 настоящих Правил) и другим компонентам, входящим в состав вакцины.
Исследования местной переносимости
31. Оценка местной переносимости осуществляется в рамках исследований по изучению общетоксического действия при однократном или повторном (многократном) введении. В случае проведения отдельных исследований по оценке местной переносимости, их необходимо проводить на животных соответствующего вида (кроликах (если не обосновано иное)). Следует использовать состав вакцины, предназначенный для клинического применения.
3.5. Оценка экологических рисков
32. Аминокислоты, пептиды, белки, углеводы и липиды, входящие в состав вакцины, не попадают под требования к проведению оценки экологических рисков, поскольку маловероятно, что они приведут к существенному риску для окружающей среды. В связи с этим для инактивированных вакцин на основании природы их компонентов оценка экологических рисков не требуется.
3.6. Дополнительные доклинические исследования отдельных видов сезонных вакцин для профилактики гриппа
Вакцины с адъювантом
33. Исследования вакцины с адъювантом проводятся с учетом требований главы 16 настоящих Правил. Должно быть дано объяснение механизма действия вакцин с адъювантом. Необходимо изучить количественные и качественные аспекты иммунного ответа. В целях получения дополнительных необходимых сведений о механизме действия адъюванта следует разработать модельные системы in vitro.
34. Методология, используемая для изучения токсичности адъювантов, должна соответствовать методологии изучения вакцины. При проведении доклинических исследований безопасности вакцин с адъювантом необходимо проводить оценку местной переносимости (реактогенности), изменения температуры тела и иммунотоксичности (например, индукции гиперчувствительности и анафилактической реакции).
35. Требования к проведению исследований иммуногенности вакцин с адъювантом на доклиническом этапе включают в себя оценку оптимального соотношения адъювант/антиген путем исследования эффекта комбинации различных доз адъюванта с различными дозами антигена вакцины.
36. Вопрос об экстраполяции данных, полученных в исследованиях на животных, требует специального рассмотрения и должен решаться с большой осторожностью. Известно, например, квасцы усиливают иммуногенность инактивированных расщепленных вакцин у мышей, хорьков и макак, однако у человека данного действия не наблюдается.
37. Необходимо отдельно изучить профиль безопасности новых адъювантов или адъювантных систем, не имеющих данных по их медицинскому применению, особенно в комбинации с новыми типами антигенов.
Живые аттенуированные сезонные вакцины для профилактики гриппа (ЖГВ)
38. В отношении живых аттенуированных сезонных вакцин для профилактики гриппа необходимо учесть следующее:
а) исследование первичной фармакодинамики. Ввиду отсутствия доказательных сведений о корреляции между системным гуморальным иммунным ответом и протективностью (защитными свойствами) ЖГВ, оценка гуморального иммунного ответа не является критерием подтверждения эффективности данного типа вакцин для профилактики гриппа. Вместе с тем исследования протективности являются научнообоснованным методом подтверждения эффективности таких вакцин, и их проведение необходимо. Исследования протективности (защитных свойств) должны подтвердить, что исследуемая вакцина способна предотвращать или существенно подавлять репликацию вируса дикого типа в тканях легкого животного и существенно снижать уровень репродукции вируса в верхних дыхательных путях.
Протективность вакцины оценивают по репродукции вируса в носовых смывах иммунизированных животных после заражения диким (патогенным) вирусом гриппа. Необходимо оценить потенциальную передачу интактным животным выделяемого вакцинного вируса;
б) фармакокинетические исследования. Необходимо проведение исследований депонирования и распределения в месте введения, а также исследований, позволяющих охарактеризовать интраназальные спреи.
Исследования должны быть проведены с использованием необходимых образцов тканей и органов с целью получения совокупности данных фармакокинетического профиля ЖГВ, кинетики распределения и оценки потенциальных рисков возникновения реверсии.
В исследованиях, как правило, достаточно использования одного вида животных, выбор которого необходимо обосновать соответствующим образом. Исследования распределения включают в себя оценку восстановления вирулентности вакцинального штамма вируса гриппа, обнаружение вирусных антигенов или вирусного генетического материала. Необходимо исключить потенциальное гематогенное распространение вакцинального вируса;
в) нейровирулентность. Необходимо изучить потенциальную нейровирулентность новых вакцинных штаммов и оценить ее в экспериментах на мышах, используя в качестве контроля вирулентный штамм;
г) токсикологические исследования. Для оценки репродуктивной токсичности (фетальной токсичности и влияние на репродуктивную функцию) вследствие возможной реверсии патогенных свойств штамма вируса гриппа особое внимание необходимо уделить выбору соответствующей модели животных (например, хорьки). Используя соответствующие модели животных (например, хорьков), необходимо изучить неблагоприятное влияние на слизистые поверхности носа, вызванное вакцинными штаммами вируса гриппа, либо вспомогательными веществами, входящими в состав препарата;
д) оценку экологических рисков. Необходимо изучить риск реассортации между диким вирусом и штаммами живых вакцинных вирусов, а также потенциальный риск распространения их среди людей и животных.
4. Требования к проведению доклинических исследований для внесения изменений в штаммовый состав вакцин для профилактики гриппа
4.1. Сезонные вакцины для профилактики гриппа
39. Представлять данные доклинических исследований при обновлении штаммов сезонных вакцин для профилактики гриппа, как правило, не требуется.
4.2. Пандемические и препандемические (зоонозные) вакцины для профилактики гриппа
40. В случае отсутствия данных по иммуногенности, полученных в ходе ранее проведенных исследований с участием человека, при подаче заявления на изменение или обновление штаммового состава вакцины для профилактики гриппа допускается представлять результаты исследований иммуногенности и протективности (защитных свойств) инактивированных вакцин, полученные в ходе исследований на животных, в соответствии с пунктами 8 - 18 настоящей главы.
41. Для живых аттенуированных вакцин для профилактики гриппа принимая во внимание отсутствие доказанной корреляции между системным гуморальным иммунным ответом и протективностью, главным критерием оценки эффективности являются исследования защитных свойств данного типа вакцины у животных.
5. Требования к проведению клинических исследований для регистрации сезонных вакцин различных типов для профилактики гриппа
5.1. Сезонные инактивированные вакцины для профилактики гриппа, не содержащие адъювант
42. При подаче заявления о регистрации новой инактивированной сезонной вакцины для профилактики гриппа не содержащей адъювант, состав и процесс производства которой аналогичен зарегистрированной инактивированной вакцине, ранее подвергшейся экспертизе уполномоченными органами (экспертными организациями) государств-членов, допускается представлять данные сравнительных исследований безопасности и иммуногенности в отдельных группах лиц, описанных в пункте 43 настоящей главы.
43. Наиболее объективную оценку эффективности получают при вакцинации лиц, не имеющих специфических антител к определенному штамму (штаммам) в составе вакцины (перед вакцинацией требуется предварительный скрининг на наличие специфических антител в сыворотках лиц, подлежащих иммунизации). Также требуются данные по иммуногенности вакцины сравнения (препарата сравнения).
44. В качестве препарата сравнения используют вакцину того же типа, вводимую тем же путем, что и исследуемая вакцина (например, субъединичная вакцина для профилактики гриппа сравнивается с зарегистрированной субъединичной вакциной, расщепленная вакцина сравнивается с зарегистрированной расщепленной вакциной).
45. Оптимальным является использование препаратов сравнения, в отношении которых получены данные, обосновывающие их эпидемиологическую эффективность (эффективность в реальных условиях применения).
46. Поскольку не существует подтвержденного иммунологического коррелята защиты, демонстрация не меньшего иммунного ответа в определенных популяциях подгрупп должна преобразовываться в максимально сопоставимую протективность против гриппозной инфекции. Граница не меньшей эффективности должна учитывать любые доступные данные по выработке естественно приобретенных антител в исследуемой популяции, а также доступную информацию по иммуногенности вакцины сравнения.
Исследования у взрослых, включая пожилых лиц
47. Для применения вакцины для профилактики гриппа у взрослых и пожилых лиц необходимо подтверждение, что вводимая разработанная вакцина обладает иммуногенностью, по крайней мере, сравнимой с иммуногенностью препарата сравнения. Если заявитель планирует провести широкомасштабные исследования в тех странах, в которых препарат сравнения не зарегистрирован, необходимо представить обоснования применимости и возможности экстраполяции данных к исследуемой популяции. Обсуждение и одобрение плана исследований с уполномоченными органами (экспертными организациями) государств-членов обязательно.
Исследования у детей
48. В связи с отсутствием данных о способности данного типа вакцин обеспечивать протективный иммунный ответ и иммунологическую память в самых младших возрастных группах, для организации исследований в детской популяции необходимо руководствоваться требованиями пунктов 49 - 53 настоящей главы. Любое отклонение от указанных требований должно быть надлежащим образом обосновано.
49. При наличии показаний, предусматривающих применение вакцины у детей в возрасте от 6 до 36 месяцев, необходимо подтвердить профилактическую (протективную) эффективность вакцины в отношении сезонного гриппа в рандомизированном клиническом исследовании.
50. При наличии показаний, предусматривающих применение вакцины у детей в возрасте от 3 до 9 лет (доля первично вакцинированных, с большой вероятностью будет вариабельной), необходимо обосновать, что иммунный ответ при выбранной дозе и схеме вакцинации будет, по крайней мере, не меньше иммунного ответа при применении вакцины у детей от 6 до 36 месяцев, для которых уже была подтверждена профилактическая эффективность. Для подтверждения сопоставимости эффективности сезонной вакцины для профилактики гриппа, не содержащей адъюванта при применении у детей в возрасте от 3 до 9 лет и при применении у детей в возрасте от 6 до 36 месяцев, необходимо проводить сравнительный анализ подмножества сывороток крови (выбранных случайным образом), полученных у детей от 6 до 36 месяцев, участвовавших в исследовании профилактической эффективности, и сывороток крови, полученных у детей от 3 до 9 лет, с использованием одного и того же метода анализа, в одной и той же лаборатории. В случаях, когда невозможно установить профилактическую эффективность вакцины при применении у детей в возрасте от 6 до 36 месяцев, заявитель в соответствии с пунктом 26 Правил регистрации и экспертизы вправе запросить консультацию у уполномоченных органов (экспертных организаций) государств-членов с целью согласования возможного объема клинических данных, которые необходимо получить для регистрации вакцины.
51. В определенных случаях допускается представлять сравнительные данные по иммуногенности (например, при подаче заявления о регистрации четырехвалентной вакцины, представляются данные, полученные для аналогичной трехвалентной вакцины, зарегистрированной для применения в данной возрастной группе). Такие данные получают:
а) в проспективном рандомизированном исследовании, направленном на сравнительный анализ иммунного ответа, индуцируемого двумя различными вакцинами в этой возрастной группе;
б) путем параллельных испытаний сывороток крови, полученных в разных исследованиях.
52. Клинические исследования вакцин данного типа с участием детей в возрасте до 9 лет проводятся в следующей хронологии:
сначала проводятся исследования безопасности и иммуногенности у детей в возрасте от 3 до 9 лет;
после подтверждения безопасности вакцины у детей старшей возрастной группы переходят к клиническим исследованиям у детей от 6 до 36 месяцев. Для детей от 6 до 36 месяцев исследования включают оценку безопасности, иммуногенности и профилактической эффективности;
после подтверждения профилактической эффективности у детей от 6 до 36 месяцев возможно скоррелировать профилактическую эффективность вакцины на группу детей от 3 до 9 лет представлением сравнительных данных по иммуногенности сывороток крови, полученных у детей от 6 до 36 месяцев и от 3 до 9 лет.
Описанная схема применима, если исследования включают одновременно детей указанных возрастных групп. Если каждая возрастная группа будет изучаться отдельно, необходимо представить результаты оценки профилактической эффективности в каждой возрастной группе.
53. В случае показаний, предусматривающих применение вакцины у детей в возрасте от 9 до 18 лет, подтверждение профилактической эффективности не требуется. При подаче заявления о регистрации представляются данные прямых сравнительных исследований иммунного ответа, индуцируемого разрабатываемой вакциной в группах лиц в возрасте от 9 до 18 лет и взрослых, либо данные сравнения с зарегистрированной инактивированной сезонной вакциной для профилактики гриппа, не содержащей адъювант, показанной к применению в данной возрастной группе. Например, допустимо сравнение новой четырехвалентной вакцины с зарегистрированной четырехвалентной вакциной. В качестве альтернативы допускается представление результатов сравнения иммунного ответа, индуцируемого разрабатываемой вакциной при исследовании с участием детей в возрасте от 9 до 18 лет и в других исследованиях с той же вакциной или с соответствующей зарегистрированной вакциной при условии проведения параллельных испытаний сывороток, полученных в разных исследованиях.
Исследования у иммунокомпрометированных лиц
54. На момент подачи заявления о регистрации проведение специальных исследований у иммунокомпрометированных лиц не требуется, за исключением случаев, когда заявитель предполагает включить показания к применению в субпопуляциях с определенным иммунным статусом.
55. Иммуногенность вакцины для профилактики гриппа будет зависеть от вида и тяжести иммунодефицитного состояния. Данные об иммуногенности, полученные в отдельных подгруппах или выбранной группе иммунокомпрометированных пациентов, позволяют вносить в общую характеристику лекарственного препарата указания, учитывающие фактически изученную популяцию. Вопрос об экстраполяции данных (например, режима дозирования) за пределы фактически изученной популяции будет определяться после полного анализа этих данных.
56. Рандомизированные контролируемые клинические исследования для оценки профилактической эффективности вакцины у иммунокомпрометированных детей проводить не требуется. Формирование группы плацебо в этой популяции неприемлемо, поскольку сложно провести научно-обоснованное исследование, а его результаты достаточно сложно интерпретировать в связи с неизбежной гетерогенностью данных для этой группы пациентов. В связи с этим для обоснования указаний применения вакцины у иммунокомпрометированных детей (начиная с минимального возраста) необходимо накапливать данные об иммуногенности у относительно небольших выборок из педиатрической популяции с различными видами и тяжестью иммунодефицитного состояния. Необходимость применения высоких доз и (или) других режимов дозирования у иммунокомпрометированных детей устанавливается путем проведения прямых и непрямых (то есть анализ данных при проведении различных исследований) сравнительных исследований между иммунокомпрометированными и сопоставимыми по возрасту здоровыми детьми.
Исследования у пациентов с сопутствующими заболеваниями
57. Результаты исследований иммуногенности у пациентов с сопутствующими заболеваниями на момент подачи заявления о регистрации вакцин для профилактики гриппа не требуются. Некоторые сопутствующие заболевания могут повысить риск осложнений гриппа, но при этом не влиять на иммунный ответ и защиту от инфекции. Эти данные получают в ходе специальных исследований у подгрупп, включенных в исследования по возрастным категориям, в которых критерии исключения минимальны. Данные об иммуногенности не прогнозируют влияние на риск осложнений у лиц, у которых, несмотря на вакцинацию, развился грипп с клиническими проявлениями. Этот риск можно оценить только во время оценки эпидемиологической эффективности вакцины в пострегистрационный период.
Исследования у беременных женщин
58. Поскольку в отношении не содержащих адъювант инактивированных сезонных вакцин (расщепленных (сплит-) и субъединичных) доступны данные об иммуногенности, безопасности и эффективности, позволяющие применять данные вакцины у беременных во всех триместрах беременности.
59. Включение в общую характеристику лекарственного препарата конкретных рекомендаций о применении новой вакцины во время беременности зависит от доступных данных, характеризующих данную вакцину. Заявителю необходимо также изучить эффективность вакцины для профилактики гриппа у детей при вакцинации матери.
5.2. Сезонные инактивированные вакцины для профилактики гриппа, содержащие адъювант
Исследования у взрослых, включая пожилых лиц
60. При подаче заявления о регистрации новой вакцины для профилактики гриппа на основе поверхностных антигенов вируса гриппа с адъювантом, которая предназначена для применения у взрослых и (или) пожилых лиц требуется представить научное обоснование включения адъюванта в состав вакцины. Таким обоснованием является демонстрация превосходства адъювантной вакцины по показателю иммуногенности в сравнении с вакциной, не содержащей адъювант, но в остальном сопоставимой, зарегистрированной вакциной, в отношении которой уполномоченными органами (экспертными организациями) государств-членов была проведена соответствующая экспертиза. Также представляется обоснование включения адъюванта в состав исследуемой вакцины, исходя из данных, подтверждающих не меньшую эффективность вакцины, содержащей адъювант по показателю иммуногенности при соответствующем снижении количества антигенов в ее составе в сравнении с вакциной, не содержащей адъювант, но в остальном сопоставимой, зарегистрированной вакциной, содержащей стандартное количество антигенов.
61. Преимущество включения адъюванта в состав вакцины заключается в повышении частоты сероконверсии, титров антител (средних геометрических титров антител или кратности приростов титров антител), или других иммунологических параметров, включая широту и длительность иммунного ответа.
62. К данным, необходимым для обоснования применения у иммунокомпрометированных лиц и людей с сопутствующими заболеваниями, применимы требования, приведенные для инактивированных вакцин, не содержащих адъювант.
Исследования у детей
63. Включение адъюванта в состав вакцины для применения у детей необходимо обосновать с представлением доказательства повышения иммунного ответа в сравнении с сопоставимой зарегистрированной вакциной, не содержащей адъювант. Также представляются данные, доказывающие не меньшую иммуногенность вакцины, содержащей адъювант со сниженным содержанием антигенов в сравнении с сопоставимой зарегистрированной вакциной, не содержащей адъювант и содержащей стандартное количество антигенов. Кроме того, подобно инактивированным вакцинам, не содержащим адъювант, применение вакцины, содержащей адъювант у детей в возрасте
 36 месяцев необходимо обосновать результатами доказанной профилактической эффективности, полученными в клинических исследованиях. Иммунологическая активность вакцины при выбранной схеме вакцинации у детей более старшего возраста, по крайней мере, не должна уступать зарегистрированной вакцине для применения у детей аналогичного возраста, для которой экспериментально подтверждена эффективность. В качестве альтернативы, при определенных обстоятельствах, заявитель вправе подать заявление о регистрации вакцины, для которой показан меньший иммунный ответ по сравнению с другой вакциной, содержащей адъювант, в отношении которой эффективность подтверждена и документирована.
36 месяцев необходимо обосновать результатами доказанной профилактической эффективности, полученными в клинических исследованиях. Иммунологическая активность вакцины при выбранной схеме вакцинации у детей более старшего возраста, по крайней мере, не должна уступать зарегистрированной вакцине для применения у детей аналогичного возраста, для которой экспериментально подтверждена эффективность. В качестве альтернативы, при определенных обстоятельствах, заявитель вправе подать заявление о регистрации вакцины, для которой показан меньший иммунный ответ по сравнению с другой вакциной, содержащей адъювант, в отношении которой эффективность подтверждена и документирована.
Исследования у беременных женщин
64. Для некоторых сезонных вакцин, содержащих адъювант проводились контролируемые клинические исследования с участием беременных женщин. Имеются данные о безопасности и эпидемиологической эффективности моновалентных пандемических вакцин, содержащих адъювант при применении во втором и третьем триместрах беременности.
65. При составлении общей характеристики лекарственного препарата необходимо учитывать все имеющиеся и значимые данные вакцинации беременных женщин. В зависимости от характеристик новой вакцины и нового адъюванта имеющиеся данные могут обосновывать или противопоставлять включение в общую характеристику лекарственного препарата рекомендации по применению вакцины во время беременности.
5.3. Сезонные живые аттенуированные вакцины для профилактики гриппа
66. Для сезонных живых аттенуированных вакцины для профилактики гриппа в связи с отсутствием доказательных сведений о корреляции между параметрами иммунного ответа и протективностью, заявитель вправе подать заявление о регистрации только на основании подтверждения профилактической эффективности вакцины в конкретных популяциях, для определенной возрастной группы.
67. При изменении лекарственной формы или средства доставки уже зарегистрированной вакцины, по предварительному согласованию с регуляторными органами, допускается проведение сравнительных исследований иммуногенности вакцин.
5.4. Прочие виды вакцин (новые вакцины)
68. При разработке новых типов вакцин для профилактики сезонного гриппа (например, рекомбинантные вакцины), для которых отсутствуют соответствующие препараты сравнения, зарегистрированные или в отношении которых проведена экспертиза уполномоченными органами (экспертными организациями) государств-членов, для подачи заявления на регистрацию требуется подтверждение профилактической эффективности разрабатываемой вакцины в клинических исследованиях в конкретных популяциях, при применении у лиц определенного возраста.
69. Заявителям необходимо получить одобрение альтернативных стратегий исследования уполномоченными органами (экспертными организациями) государств-членов на ранних этапах разработки протокола клинических исследований (например, для согласования возможного подтверждения эффективности в определенных возрастных и популяционных подгруппах с последующей экстраполяцией на остальные группы, основываясь на полученных данных иммуногенности).
6. Требования к проведению клинических исследований для внесения изменений в штаммовый состав вакцин для профилактики гриппа
70. В связи с потенциалом изменчивости вирусов гриппа и циркуляцией эпидемически активных штаммов, ВОЗ дважды в год (в феврале для северного полушария и в сентябре для южного полушария), обновляет рекомендации в отношении сезонной вакцины для профилактики гриппа (для каждого из полушарий).
71. На основании этой рекомендации замена штаммового состава в зарегистрированных вакцинах осуществляется посредством внесения изменений в их регистрационное досье (в соответствии с приложением № 24 к Правилам регистрации и экспертизы).
72. Допускается не представлять данные клинических исследований при изменении или обновлении штаммов сезонных вакцин для профилактики гриппа. Вместе с тем необходимо обеспечить пострегистрационные наблюдения посредством проведения мониторинга безопасности и эпидемиологической эффективности такой вакцины (в соответствии с Правилами практики фармаконадзора, пунктами 130 - 144 и разделом 10 настоящей главы).
7. Требования к проведению доклинических и клинических исследований для регистрации пандемических вакцин для профилактики гриппа и внесения изменений в их штаммовый состав
73. В целях подготовки к пандемии производители вакцин подают заявление о регистрации пандемической вакцины (вакцину готовности к пандемии). При официальном признании пандемии (объявления ВОЗ в установленном порядке пандемической ситуации или объявления соответствующими уполномоченными органами (экспертными организациями) государств-членов эпидемии, вызванной пандемическим штаммом вируса гриппа) заявитель вправе представить заявление на изменение штаммового состава с целью включения штамма с пандемическим потенциалом в пандемическую вакцину («обновление пандемического штамма»). Заявление о регистрации вакцины готовности к пандемии необходимо обосновать данными о соответствующих штаммах (штамме) (в соответствии с приложением № 24 к Правилам регистрации и экспертизы).
7.1. Требования к исследованиям вакцины готовности к пандемии в целях ее регистрации
74. Заявление о регистрации вакцины готовности к пандемии должно содержать данные с аналогичными характеристиками, что и планируемая для применения вакцина против пандемического штамма вируса гриппа, а именно, единую технологию приготовления, состав вакцины (за исключением штамма (штаммов)), в частности, содержание антигена, вспомогательных веществ, адъюванта (в случае необходимости) и др., а также показатели, заложенные в спецификацию вакцины и методы контроля качества. Регистрационное досье должно содержать данные по безопасности и иммуногенности вакцины, содержащей штамм с пандемическим потенциалом, со слабовыраженными иммуногенными свойствами и к которому большое количество людей не имеет специфического иммунитета (вследствие отсутствия перенесения такого гриппа в анамнезе; например, H5N1). Эта стратегия позволяет определить режим дозирования, который, скорее всего, окажется схожим, если очередная пандемия будет вызвана этим штаммом. Данные по безопасности и иммуногенности такой же вакцинной конструкции, содержащей другие потенциальные пандемические штаммы и сезонные штаммы, включаются в регистрационное досье в качестве дополнительного подтверждения (если эти данные значимы для принятия решения о регистрации вакцины).
Инактивированные вакцины готовности к пандемии
75. Для подачи заявления о регистрации инактивированной вакцины готовности к пандемии необходимо представить данные по безопасности и иммуногенности, полученные в соответствии с требованиями, описанными в пункте 74 настоящей главы.
76. Для оценки эффективности вакцины в случае возникновения пандемии, требуется проведение исследований двух или более образцов вакцины одной и той же конструкции (состава), содержащих слабоиммуногенные штаммы, которыми большинство людей не инфицировалось. В регистрационное досье необходимо включить все полученные ранее данные по безопасности и эффективности в отношении той же или сходной (в зависимости от особенностей производственных процессов) конструкции (состава) вакцины, зарегистрированной и подвергшейся экспертизе уполномоченными органами (экспертными организациями) государств-членов (например, сезонные или препандемические вакцины).
77. Регистрационное досье должно содержать данные по безопасности и иммуногенности, полученные при применении у здоровых взрослых в возрасте 18 лет и старше, включая лиц, начиная с 60 лет и старше. В зависимости от степени риска (насколько это возможно), необходимо получить данные по безопасности и иммуногенности вакцины при применении у здоровых детей (данные применения у детей также допускается получать, используя результаты применения препандемической вакцины, если она зарегистрирована). Исследования по применению моновалентных пандемических вакцин с адъювантом (например, расщепленной и субъединичной вакцины с антигенным составом HINlv) у беременных подтверждают иммуногенность и эпидемиологическую эффективность, а также безопасность, что может служить обоснованием применения этих вакцин во всех триместрах беременности. Тем не менее, при составлении общей характеристики лекарственного препарата необходимо учитывать все доступные и значимые данные применения у беременных.
78. В ходе пандемии необходимо оценить эпидемиологическую эффективность вакцины в соответствии с представленным планом управления рисками (в соответствии с разделом 10 настоящей главы). Во время пандемии пострегистрационные исследования должны включать в себя мониторинг безопасности и эпидемиологической эффективности в популяциях, включенных и не включенных в исследования безопасности и иммуногенности, предусмотренных в регистрационном досье.
Живые аттенуированные вакцины готовности к пандемии
79. Известно, что показатели гуморального системного иммунного ответа на введение ЖГВ не коррелируют с протективностью вакцины. Тем не менее при выборе режимов дозирования ЖГВ, содержащей штаммы вируса гриппа с пандемическим потенциалом, возможно проведение исследований у лиц, ранее не имевших случаев инфицирования выбранным вакцинным штаммом вируса гриппа по следующей схеме: однократная доза ЖГВ с последующим (спустя определенный срок) введением дозы инактивированной вакцины не содержащей адъювант, содержащей тот же штамм. Результаты иммунного ответа на первую и вторую дозы предоставляют информацию о способности однократной дозы ЖГВ праймировать различные возрастные группы против слабоиммуногенного штамма, к которому большинство, если не все, не имеют специфического иммунитета. Такой дизайн исследования признается обоснованным в качестве косвенного подтверждения потенциальной протективности пандемической вакцины ЖГВ при отсутствии данных об эффективности в период между пандемиями, однако его не следует расценивать в качестве образцового режима дозирования ЖГВ в пандемических условиях. Подход к выбору штаммов аналогичен подходу к выбору штаммов инактивированных вакцин.
80. Лица, участвующие в клинических исследованиях ЖГВ в период между пандемиями или в фазу пандемической тревоги, должны находиться в соответствующих условиях клинической изоляции. Такие исследования с участием детей в период между пандемиями недопустимы.
81. Данные о профилактической и (или) эпидемиологической эффективности, полученные в отношении ЖГВ, содержащей эпидемически актуальные штаммы, можно рассматривать в качестве дополнительных сведений в отношении той же конструкции (состава), содержащей штамм с пандемическим потенциалом. Требования к пострегистрационным исследованиям этих вакцин аналогичны требованиям для пострегистрационных исследований для инактивированных вакцин готовности к пандемии.
7.2. Требования к исследованиям вакцины готовности к пандемии в целях внесения изменения в ее штаммовый состав
82. После официального объявления ВОЗ о пандемии и объявления соответствующими уполномоченными органами государств-членов эпидемии, вызванной пандемическим типом вируса гриппа заявитель вправе представить в уполномоченный орган (экспертную организацию) референтного государства заявление о внесении изменений в состав пандемических вакцин для профилактики гриппа.
83. Регистрационное досье пандемических вакцин для профилактики гриппа составляется на основе неполных данных и содержит данные о качестве, имеющиеся данные клинических исследований, свидетельствующих о предполагаемой иммуногенности штамма с пандемическим потенциалом. Если представление таких данных клинических исследований невозможно, то в дальнейшем, после объявления пандемии, заявитель должен представить в уполномоченный орган (экспертную организацию) референтного государства недостающие данные клинических исследований, а в регистрационное досье включить соответствующее обоснование, привести описание недостающих данных и гарантировать их представление в будущем. В то же время необходимо активировать планы по оценке эпидемиологической эффективности вакцины, а результаты представить в срок, согласованный заявителем и уполномоченным органом (экспертной организацией) референтного государства.
84. В случае необходимости внесения изменений штаммового состава пандемических вакцин для профилактики гриппа в периоды между пандемиями, заявитель вправе обратиться к уполномоченным органам (экспертным организациям) государств-членов за консультацией относительно требований к представляемым данным.
7.3. Требования к проведению исследований при экстренной процедуре регистрации вакцин для профилактики гриппа в условиях пандемии
85. При объявлении ВОЗ в установленном порядке пандемической ситуации или объявления соответствующими уполномоченными органами государств-членов эпидемии, вызванной пандемическим типом вируса гриппа, регистрация новой пандемической вакцины осуществляется на основании экстренной процедуры. Если планируется экстренная процедура, необходимо как можно раньше инициировать обсуждение такой процедуры с уполномоченными органами (экспертными организациями) государств-членов.
86. Совокупность данных, требуемых для регистрации инактивированных и живых аттенуированных вакцин для профилактики гриппа, будет варьировать в зависимости от типа вакцины, и учитывать все доступные сведения, для каждой конкретной технологии производства вакцин. В случае регистрации новой технологии производства вакцин, для обоснования возможности регистрации вакцины следует представить большее количество данных по сравнению с вакциной хорошо изученной конструкции.
8. Требования к проведению доклинических и клинических исследований для регистрации препандемических вакцин для профилактики гриппа и внесения изменений в их штаммовый состав
8.1. Требования к исследованиям препандемической (зоонозной) вакцины для профилактики гриппа в целях ее регистрации
87. Заявление о регистрации препандемической (зоонозной) вакцины для профилактики гриппа должно содержать данные применения такой вакцины в конкретной популяции. Например, если препандемическая вакцина, содержащая штамм A/Indonesia/05/2005 (H5N1), исследована у взрослых, то в показаниях к применению для профилактики гриппа, вызываемого A/Indonesia/05/2005 (H5N1), должно быть указано «применение только у взрослых». Заявители вправе представить дополнительные сведения в отношении той же технологии производства вакцины, но содержащей другие зоонозные штаммы.
88. На момент подачи заявления о регистрации препандемических вакцин данные профилактической эффективности представлять не требуется. Вместе с тем в случае применения вакцины во время вспышек инфекции, вызванной зоонозными штаммами вируса гриппа необходимо накапливать сведения об эффективности и безопасности применяемой вакцины. Для каждой вакцинированной группы вне зависимости от когорты субъектов исследования необходимо провести оценку иммунного ответа.
89. В заявление о регистрации вакцины должны быть включены данные о специфических антителах и иммунном ответе на введение бустерных доз в когортах каждой возрастной группы и групп риска, которые планируется отразить в показаниях к применению. В случае отсутствия в регистрационном досье таких данных необходимо проведение пострегистрационных исследований.
8.2. Требования к исследованиям препандемической (зоонозной) вакцины для профилактики гриппа в целях внесения изменений в ее штаммовый состав
90. В случае появления данных, свидетельствующих о низкой кросс-реактивности (перекрестная иммуногенность) и кросс-протективности (перекрестная защита) против дрейфовых вариантов вируса необходимо внести изменения в штаммовый состав препандемической (зоонозной) вакцины. В зависимости от типа замены штамма вируса гриппа зоонозного происхождения требования о внесении изменений в регистрационное досье будут отличаться:
в случае изменения (обновления) штамма вируса гриппа зоонозного происхождения зарегистрированной вакцины другим штаммом того же подтипа (серотипа) (например, замена референс-штамма H5N1 штаммом H5N1 определенного клайда). В этом случае, при соответствующем обосновании, держатель регистрационного удостоверения вправе представить заявление о внесении изменений в регистрационное досье, содержащее только данные о производстве и качестве, касающиеся нового штамма (в соответствии с приложением № 24 к Правилам регистрации и экспертизы). Для оценки кросс-реактивности необходимо провести вакцинацию пациентов, ранее получавших зарегистрированную вакцину, вакциной с измененным штаммовым составом. Однако такие данные представляются только после согласования заявления о внесении изменений в штаммовый состав вакцины для профилактики гриппа зоонозного происхождения с уполномоченными органами (экспертными организациями) государств-членов:
в случае изменения (обновления) штамма вируса гриппа зоонозного происхождения зарегистрированной вакцины штаммом другого подтипа (серотипа), отличающегося по гемагглютинину (НА) и нейраминидазе (NA) (например, изменение (обновление) исходного штамма H5N1 другим штаммом, например, H7N7) необходимо провести оценку иммуногенности и безопасности, и провести консультации с уполномоченными органами (экспертными организациями) государств-членов.
9. Организационные, методическое и научные аспекты проведения клинических исследований вакцин для профилактики гриппа
91. Требуется согласование программы клинических исследований с уполномоченными органами (экспертными организациями) государств-членов.
92. Объем клинических данных, выбор методов количественного определения параметров, используемых для интерпретации данных, должны быть обоснованы.
9.1. Организация и проведение клинических исследований оценки иммуногенности вакцин для профилактики гриппа
93. Оценка иммуногенности вакцин для профилактики гриппа основывается на применении двух методов: реакции торможения гемагглютинации (РТГА), обнаруживающем сывороточные антигемагглютинирующие антитела, и реакции одиночного радиального гемолиза (РРГ). Ни один из этих методов не стандартизирован. Показано, что результаты, получаемые в разных лабораториях с использованием этих методов, достаточно вариабельны. Программа клинических исследований должна предусматривать проведение оценки иммуногенности с использованием методов реакции торможения гемагглютинации и реакции одиночного радиального гемолиза в надлежащим образом аттестованных лабораториях. Все испытания необходимо проводить в одной или ограниченном количестве лабораторий, используя одинаковую методику анализа. Необходимо хранить сыворотки в течение длительного времени, чтобы можно было провести повторные испытания по мере совершенствования методов испытаний (например, они могут подвергнуться повторным испытаниям в рамках процесса валидации). Заявители должны использовать валидированные методы и собственные контроля, унифицированные лабораторные протоколы и стандартные реактивы. Необходимо использовать международные стандарты вакцинных антигенов при их наличии.
94. Реакция нейтрализации вирусов (PH) позволяет количественно определять вируснейтрализующие антитела. Метод основан на обнаружении способности сывороток вакцинированных людей в различных разведениях блокировать репликацию вирусов при культивировании в клеточной культуре MDCK (в случае использования в реакции микропланшета - реакция микронейтрализации (PH)). Титр вируснейтрализующих антител необходимо определять в каждом исследовании, по крайней мере, у репрезентативной подгруппы исследуемой популяции, но предпочтительным является проведение анализа сывороток всех включенных в исследования лиц. Вместе с тем, аналогично реакции торможения гемагглютинации и реакции одиночного радиального гемолиза (РРГ), метод не стандартизирован, кроме того, в настоящее время имеется лишь ограниченное количество данных, что не всегда позволяет применять конкретный метод при оценке иммунологических данных. К критичным параметрам метода, оказывающим влияние на результаты, относятся:
способ учета данных;
продолжительность инкубации;
использование трипсина.
Для получения достоверных сведений требуется обоснование выбора используемой методологии и изучения ее влияния на получаемые результаты. Первое разведение образца сыворотки не должно превышать 1:10. На протяжении всей программы клинических исследований необходимо использовать одинаковую аналитическую методологию и проводить исследования в одной и той же аттестованной лаборатории.
95. Необходимо проводить оценку клеточного иммунитета, по крайней мере, у случайно выбранных подгрупп во всем целевом диапазоне возрастов. Оценка клеточного иммунного ответа у пожилых лиц (например, в возрасте 75 лет и старше) особенно информативна в связи с тем, что антитела, определяемые в реакции торможения гемагглютинации и реакции нейтрализации вирусов, превышающие такие же значения у взрослых лиц молодого возраста, могут не спрогнозировать протективность вакцины.
96. В исследованиях оцениваются количественные и качественные показатели Т-клеточного иммунного ответа. Например, необходимо проводить оценку антиген-специфичных Т-клеток (таких как Th1, Th2, регуляторные Т-клетки, Т-клетки памяти и соответствующие цитокины). Кроме того, детальный анализ CD4+- и CD8+-T - лимфоцитов, а также активации В-клеток памяти позволит лучше охарактеризовать влияние вакцинации на гуморальный иммунный ответ и протективность.
97. Допускается также проведение заявителями оценки антинейраминидазных (NA) антител, по крайней мере, у случайно выбранных подгрупп. В случае проведения клинических исследований оценки иммуногенности метод количественного определения должен быть валидирован и проводиться в аттестованных лабораториях.
98. Заявители также оценивают кинетику антителообразования как показателя состояния после первичной иммунизации и формирования иммунного ответа. Такие данные должны быть документированы и информативны в исследованиях, проводимых при вакцинации первично неиммунизированных лиц.
99. В связи с патогенностью и эпидемиологическими особенностями штаммов вируса гриппа зоонозного происхождения, необходимо провести оценку сывороток крови, полученных от лиц, вакцинированных препандемическими (зоонозными) и пандемическими вакцинами для определения следующих показателей:
кросс-реактивности - перекрестного иммунного ответа на выбранный вакцинный штамм к дрейфовым вариантам того же подтипа вируса гриппа (например H5N1) (исследования in vitro);
кросс-праймирования - сравнительные данные результатов анамнестического иммунного ответа на повторное введение не дрейфового штамма, но близкого к нему после первичной вакцинации вакцинным штаммом, и на первую дозу дрейфового штамма у ранее невакцинированной контрольной группы;
кросс-протективности (перекрестной защиты от инфекции) - если представляются данные о наличии кросс-протективности, они должны основываться на демонстрации перекрестного иммунного ответа в сыворотках, полученных от вакцинированных лиц, подкрепленные доклиническими данными.
100. Ввиду непрерывного штаммового дрейфа, заявителям необходимо после первичной регистрации проводить дальнейшее исследование вакцин по указанным параметрам.
101. Протоколы исследования должны содержать подробное описание методологий, применяемых для оценки иммунного ответа вакцинированных, а также обоснование режима взятия образцов. Если в ходе разработки программы клинических исследований требуется внести изменения, необходимо представить соответствующее обоснование.
9.2. Анализ и представление иммунологических данных
Представление данных для всех типов вакцин для профилактики гриппа
102. Данные по иммуногенности вакцин для профилактики гриппа, полученные с использованием методов реакции торможения гемагглютинации (и (или) реакции одиночного радиального гемолиза) и реакции нейтрализации вирусов, необходимо подробно представить для каждого штамма, входящего в состав вакцины, используя при формировании каждого отчета об исследовании стандартный подход. Как минимум для результатов каждого проведенного анализа необходимо представить следующие данные:
средние геометрические титры антител (с доверительным интервалом 95%) и кратность приростов титров антител (соотношение превакцинных и поствакцинальных средних геометрических титров антител);
кривые обратного кумулятивного распределения, которые необходимо сопроводить таблицами с данными о доле вакцинированных лиц (в процентах), титры которых превышают определенный уровень титров на логарифмической кривой (например, титры превышающие 1:10, 1:100 и 1:1 000);
фактор и уровень сероконверсии. Сероконверсию допускается определить различными способами, включая, кратность нарастания титра по сравнению с исходным уровнем и (или) появление определяемого титра у субъекта исследования с ранее не обнаруживаемыми или количественно не определяемыми антителами;
анализ подгрупп исследуемой популяции по таким факторам, как возраст и предшествующий иммунный статус;
данные об иммунологическом ответе на ревакцинацию, если она предусмотрена и необходима, основываясь на иммунологическом статусе до введения дополнительной дозы;
данные об антиген-специфичном Т-клеточном ответе, включая CD4+ Т-клетки и СЭ8+-цитотоксические Т-лимфоциты (ЦТЛ) и соответствующие цитокины, с учетом их исходного уровня.
В случае если в каком-либо испытании используется несколько штаммов данные должны быть представлены для каждого штамма по отдельности.
Представление данных для пандемических и препандемических (зоонозных) вакцин для профилактики гриппа
103. С использованием методов реакции торможения гемагглютинации и (или) реакции одиночного радиального гемолиза, а также реакции нейтрализации вирусов необходимо представить доказательные сведения об иммуногенной активности вакцины. Необходимо провести оценку доли вакцинированных лиц с высокими титрами антител (значения в пределах доверительного интервала 95%), а также фактор (кратность увеличения средних геометрических титров антител) и уровень сероконверсии. В целях описания корреляции необходимо сравнить данные, полученные с использованием различных методов: (реакции торможения гемагглютинации и (или) реакции одиночного радиального гемолиза, а также реакции нейтрализации вирусов).
104. Необходимо представить данные о кросс-реактивности и кросс-праймировании (как описано в пункте 99 настоящей главы).
9.3. Виды исследований иммуногенности
Исследования подбора дозы
105. Регистрационное досье гриппозной вакцины должно содержать данные, обосновывающие выбранную дозу, режим вакцинации и состав вакцины для различных целевых групп, которые планируется включить в показания к применению (распределение по возрасту и состоянию здоровья). Любое отклонение от требований должно быть обосновано и согласовано с уполномоченными органами (экспертными организациями) государств-членов.
106. Включение адъюванта в состав вакцины должно быть обосновано. В обосновании необходимо представить доказательные сведения о повышении иммунного ответа при добавлении адъюванта, потенциально приводящего к возможному снижению содержания антигена, и приемлемом профиле безопасности. Необходимо представить доказательные сведения, что выбранное для исследования соотношение количества антиген-адъювант является оптимальным для развития иммунного ответа на антиген с минимальным риском нежелательных явлений.
107. Необходимо провести исследование режимов дозирования вакцин с адъювантом, предназначенных для применения у детей и пожилых.
108. Если заявитель разрабатывает инактивированную сезонную вакцину без адъюванта для применения у детей, перед принятием решения о проведении исследований эффективности необходимо получить данные об иммунном ответе и провести соответствующие исследования по подбору дозы. Исследования по подбору дозы у детей необходимо проводить и оценивать подобно другим возрастным группам. Схемы первичной иммунизации необходимо изучить в группе детей в возрасте от 6 до 36 месяцев, которые наиболее вероятно не будут иметь специфического иммунитета к гриппу. В случае если вакцина оказывается слабоиммуногенной при применении у детей в возрасте  36 месяцев (то есть с выраженным низким иммунным ответом по сравнению с группой детей старшего возраста или подростками), проведение оценки профилактической эффективности вакцины в этой возрастной группе может быть нецелесообразным. Если результаты исследования по подбору дозы свидетельствует о том, что иммунный ответ на отдельную вакцинную конструкцию (состав) существенно отличается для одного из штаммов сезонной вакцины или на отдельный штамм, включенный в пандемическую или препандемическую (зоонозную) вакцину, такие результаты перед продолжением программы клинической разработки необходимо обсудить с уполномоченными органами (экспертными организациями) государств-членов.
36 месяцев (то есть с выраженным низким иммунным ответом по сравнению с группой детей старшего возраста или подростками), проведение оценки профилактической эффективности вакцины в этой возрастной группе может быть нецелесообразным. Если результаты исследования по подбору дозы свидетельствует о том, что иммунный ответ на отдельную вакцинную конструкцию (состав) существенно отличается для одного из штаммов сезонной вакцины или на отдельный штамм, включенный в пандемическую или препандемическую (зоонозную) вакцину, такие результаты перед продолжением программы клинической разработки необходимо обсудить с уполномоченными органами (экспертными организациями) государств-членов.
Исследования иммунологической памяти и необходимости в ревакцинации
109. На момент регистрации сезонных вакцин для профилактики гриппа необходимо определить и обсудить с уполномоченными органами (экспертными организациями) государств-членов необходимость оценки иммунного ответа на введение бустерных доз. Данные целесообразно получить до подачи заявления на регистрацию или после регистрации в следующих случаях:
сезонные вакцины содержащие адъювант. Для того чтобы оценить необходимость ежегодной ревакцинации, в отношении сезонных вакцин, содержащих адъювант, необходимо изучить устойчивость иммунного ответа на первичную вакцинацию до 12 месяцев после окончания вакцинации (то есть до начала введения вакцины следующего сезона). Если есть популяции, для которых ежегодная вакцинация не рекомендована, устойчивость антител допускается изучать свыше 12 месяцев. В таких популяциях заявители должны предусмотреть подгруппы участников исследования для бустерного введения вакцины через один и два года после первичной иммунизации в целях изучения влияния потребности в повторной дозе и сроков ее введения;
инактивированные пандемические и препандемические (зоонозные) вакцины. В отношении пандемических и препандемических (зоонозных) вакцин данные об их иммуногенности необходимо накапливать в течение 6 месяцев после первичной вакцинации с целью оценки иммунологической памяти и (или) необходимости в ревакцинации (если применимо).
Исследования оценки иммунологических коррелятов защиты
110. При оценке эффективности инактивированных вакцин для профилактики гриппа исходят из предположения, что титр сывороточных антигемагглютинирующих антител в реакции торможения гемагглютинации, равный 1:40, обеспечивает 50 - 70% защиту от инфекции у здоровых взрослых. При этом в настоящее время появляется все больше данных, свидетельствующих о том, что требуются более доказательные сведения о корреляции иммунологической активности и защитного эффекта, которые в свою очередь, могут варьировать в зависимости от типов вакцины, исследуемой популяции, возрастной группы (например, дети, пожилые лица).
111. В ходе программ разработки клинических исследований новых вакцин для профилактики гриппа заявители должны получить данные, которые могут выявить корреляты протективности от клинически манифестируемого гриппа. В ходе исследований эффективности необходимо изучить различные параметры иммунного ответа, а также провести анализ изучения корреляции между параметрами иммунного ответа и защитой от инфекции.
9.4. Исследования профилактической (протективной) эффективности
112. В настоящем разделе рассматривается дизайн клинических исследований профилактической (протективной) эффективности вакцин для профилактики гриппа в случаях, когда проведение таких исследований необходимо и целесообразно.
Дизайн исследования и выбор контроля
113. Клинические исследования должны быть проспективными рандомизированными контролируемыми двойными слепыми. Клинические исследования вакцин, как правило, проводятся с целью подтверждения эффективности вакцинации в сравнении с невакцинированной группой. Для обеспечения проведения двойного слепого метода клинического исследования при оценке эффективности вакцины целесообразно использовать вместо плацебо по возможности негриппозную контрольную вакцину (например, вакцины, предназначенные для профилактики других заболеваний). В качестве альтернативного варианта при достаточном обосновании заявители вправе провести исследование с активным контролем, в котором контрольной вакциной будет являться зарегистрированная вакцина против гриппа. В этом случае целью исследования будет являться подтверждение превосходства исследуемой вакцины над зарегистрированной (например, превосходство вакцины, содержащей адъювант, над вакциной, не содержащей адъювант). В зависимости от характеристик исследуемой вакцины и выбранного контроля, а также при достаточном обосновании необходимо предусмотреть анализ первичного конечного показателя исходя из подтверждения не меньшей эффективности. Выбор границ не меньшей эффективности подлежит соответствующему обоснованию заявителем.
114. При рандомизации необходимо учесть вероятность систематической ошибки. Количество субъектов исследования в каждом клиническом исследовании должно быть достаточным для обеспечения достижения целей исследований. Критерии невключения в исследование должны быть минимальными. В целях обеспечения адекватной оценки профилактической эффективности вакцины для профилактики гриппа при формировании основной и контрольной групп за единицу выборки необходимо принимать группу лиц, находящихся в одних и тех же условиях, например, по возрастным категориям (дети, взрослые, пожилые) или иным признакам (пациенты с сопутствующими заболеваниями), поскольку ожидается разный иммунный ответ на вакцину.
115. При определении единицы выборки при проведении исследований с участием детей необходимо учитывать возраст и иммунологический статус к вирусу гриппа, поскольку большинство субъектов исследования не будут иметь специфического иммунитета к гриппу.
116. При проведении исследований в группе пожилых необходимо предусмотреть, чтобы исследуемая популяция включала лица, как проживающие дома, но получающие социальные услуги на дому, так и проживающие в домах престарелых. В репрезентативную выборку необходимо включить лиц старше 75 лет.
117. Протоколы исследований защитной эффективности должны заранее определять оптимальные сроки и подгруппы взятия образцов с целью оценки показателей иммуногенности, а также оговаривать используемые методы. Если иммунологическому исследованию будут подвергаться сыворотки из подгруппы популяции, процесс отбора должен обеспечивать широкую репрезентативность образцов общей исследуемой популяции.
Первичные и вторичные конечные точки
118. Для установления эффективности исследуемой вакцины для профилактики гриппа в качестве основы первичной конечной точки должны быть приняты все случаи гриппоподобных заболеваний (ГПЗ), подтвержденные с использованием лабораторных методов, применяемых для диагностики гриппа (метод полимеразной цепной реакции или культуральный метод (выделение вирусов гриппа в клеточных культурах и куриных эмбрионах)). В случае если заявитель в качестве первичной переменной планирует использовать альтернативные или дополнительные показатели, то перед началом исследования эффективности их необходимо согласовать с уполномоченными органами (экспертными организациями) государств-членов. Например, при проведении исследований в группе пожилых лиц с лабораторно подтвержденным гриппом, в качестве первичной конечной точки может рассматриваться составная конечная точка, включающая случаи грипп-ассоциированной пневмонии, госпитализацию и смертность, вызванную гриппом.
119. При планировании исследований исходят из предположения о том, что в подавляющем большинстве зафиксированных случаев в исследовании будет преобладать один штамм или серотип (например, А/Н1N1/ или A/H3N2/ или определенная линия вируса гриппа типа В), даже при проведении исследований на протяжении нескольких сезонов.
120. В зависимости от штамма, фактически преобладающего в зафиксированных случаях, заявители должны включить в обсуждение ожидаемую эффективность в отношении других типов вируса гриппа (например, при исследовании эффективности в отношении вируса гриппа серотипа A(H1N1), заявитель должен обсудить возможную экстраполяцию данных эффективности в отношении A(H3N2)). Кроме того, необходимо провести пострегистрационные исследования по оценке эпидемиологической эффективности в отношении специфичного штамма.
121. Критичной вторичной конечной точкой является оценка эффективности исследуемой вакцины против гриппа, вызванного штаммами, которые соотносятся со штаммами входящими в состав вакцины. Если исследования эффективности проводятся в сезон, когда рекомендуемые вакцинные штаммы не соответствуют преобладающим циркулирующим штаммам, это может повлиять на оценку эффективности вакцины, основанную на рекомендованной первичной конечной точке. В таком случае определение эффективности вакцины для профилактики гриппа для случаев, вызванных хорошо соответствующими штаммами, будет представлять значительную важность при оценке общей потенциальной пользы вакцины, при этом количество случаев, вызванных данными штаммами, во время исследования эффективности должно быть достаточным для проведения указанной оценки.
122. Другие вторичные конечные точки должны включать все случаи смертности, госпитализации, гриппоподобных симптомов, все случаи грипп-ассоциированной пневмонии и для детей - средний отит. Если оценивается снижение показателя вторичной очаговости в домашних или в школьных условиях, данные должны основываться на лабораторно подтвержденных случаях гриппа.
123. В протоколе исследований необходимо заранее описать анализ вторичных конечных точек, направленный на оценку частоты возникновения гриппа с учетом вакцинации препаратами для профилактики гриппа, поскольку для адекватной оценки эффективности вакцин для профилактики гриппа и получения достоверных данных о первичной иммунизации необходимо, прежде всего, учитывать исходный серологический статус субъектов исследования.
124. Заявитель вправе изучать конечные точки, касающиеся образа жизни (невыход на работу, использование ресурсов здравоохранения, затраты), однако эти данные не являются обоснованием достигнутой профилактической (протективной) эффективности при оценке эффективности вакцины.
Продолжительность исследования
125. В связи с неопределенностью продолжительности эпидемий и вероятности совпадения циркулирующих эпидемических и вакцинных штаммов в конкретном эпидсезоне сложно заранее определить необходимое количество сезонов, подлежащих включению в рамках одного клинического исследования. Для более надежной оценки эффективности вакцины клинические исследования требуют включения в исследования нескольких сезонов вакцинации.
126. Если исследование проведено в популяции, требующей ежегодной ревакцинации, то оценку результатов такого исследования необходимо включить в протокол исследования и план статистического анализа. Если исследование проводится в подгруппе популяции и странах, в которых вакцинация конкретно этой группы не рекомендуется, то данные, полученные во втором сезоне после вакцинации, потенциально информативны относительно протективности и кросс-протективности.
127. Поскольку в настоящее время получены лишь ограниченные данные эффективности вакцин содержащих адъювант и живых аттенуированных вакцин для профилактики гриппа в течение нескольких сезонов, вопрос о необходимости ревакцинации в последующих сезонах необходимо изучить в клинических исследованиях.
9.5. Исследования эпидемиологической эффективности (эффективность в реальных условиях применения) вакцин для профилактики гриппа
128. В план управления рисками необходимо включить пострегистрационные исследования эпидемиологической эффективности в отношении всех сезонных и пандемических вакцин для профилактики гриппа, включая вакцины, зарегистрированные в настоящее время, и новые вакцины.
129. Проведение надлежащей оценки эпидемиологической эффективности не всегда является возможным, поэтому держатель регистрационного удостоверения должен представить обоснование отсутствия или ограниченности данных в конкретный эпидемический сезон. При обосновании допускается представлять результаты метаанализа оценки эпидемиологической эффективности отдельной вакцины на протяжении нескольких сезонов. Пострегистрационные исследования необходимо планировать и проводить на заранее определенных географических территориях в соответствии со специально разработанной программой.
130. В пунктах 131 - 136 настоящей главы приведены указания по дизайну исследований по оценке эпидемиологической эффективности сезонных-вакцин для профилактики гриппа и пандемических вакцин в условиях пандемической ситуации.
Принципы планирования исследования
131. Для оценки эпидемиологической эффективности вакцин для профилактики гриппа необходимо использовать протоколы исследований, ранее использовавшиеся в условиях сезонных эпидемий и пандемии, например протокол исследования «случай - контроль» или проспективного когортного исследования, основанного на популяционных базах данных (регистрах), например, с валидацией клинических исходов с помощью полимезарной цепной реакции в подгруппе субъектов исследования. В случае, когда это невыполнимо, эпидемиологическая эффективность вакцины для профилактики гриппа оценивается по соотношению лабораторно подтвержденных случаев заболевания вакцинированных лиц к общей вакцинированной популяции. Для оценки эпидемиологической эффективности вакцины против гриппа необходимо провести исследования «случай - контроль» по сравнению с лабораторно подтвержденным гриппом.
Конечные точки и критерии признания случая
132. В каждом дизайне исследования необходимо предусмотреть лабораторно подтвержденные случаи гриппа. Выбор конечных точек исследования зависит от дизайна исследования.
133. Для дизайна «случай - контроль» или дизайна «случай - контроль с отрицательным результатом» первичной конечной точкой исследований должен быть случай лабораторно подтвержденного гриппа. На основании условий исследования (общая популяция или стационары) вторичные конечные точки могут учитывать способность вакцин предотвращать пневмонию и госпитализацию (обусловленные гриппом или связанные с ним респираторные или кардиологические нарушения), либо смерть.
134. Для когортного дизайна возможными конечными точками являются:
респираторные инфекции, требующие оказания медицинской помощи;
гриппоподобные заболевания, требующие оказания медицинской помощи;
все случаи смертности;
смертность от респираторных инфекций;
госпитализация в связи с пневмонией и гриппом;
все случаи госпитализации респираторных заболеваний;
лабораторно подтвержденные случаи респираторных инфекций и (или) пневмонии и гриппа с госпитализацией;
поступление пациента в отделение интенсивной терапии.
135. В дополнение к оценке эпидемиологической эффективности вакцины держателям регистрационного удостоверения необходимо в рамках тех же исследований эпидемиологической эффективности осуществлять анализ образцов на наличие антигена или выделение вируса из образцов.
Необходимо фиксировать возраст субъектов исследования, вакцинальный статус (включая анамнез вакцинации), тяжесть заболевания у вакцинированных лиц, географический район и неделю начала гриппоподобных заболеваний в течение сезона или пандемии. Эти данные важны при рассмотрении данных об эпидемиологической эффективности вакцины (например, соотнесение эффективности вакцины с циркулирующими штаммами, эффективности вакцины в отношении дрейфовых вариантов вируса гриппа в течение эпидемического сезона или пандемии).
136. Критерием признания случая гриппа является лабораторное подтверждение гриппа методом ПЦР с обратной транскрипцией (ОТ-ПЦР) или с использованием культуры клеток в аттестованных лабораториях.
Целевая популяция
137. Эпидемиологическую эффективность вакцины для профилактики гриппа необходимо исследовать в популяции, которая указана в показаниях к применению такой вакцины. Данные допускается получать лишь в той стране, в которой вакцина зарегистрирована.
138. Необходимо сопоставлять данные возрастных когорт:
детей, подростков;
лиц младше 65 лет;
лиц в возрасте 65 лет и старше;
лиц старше 75 лет.
139. Необходимо оценить эпидемиологическую эффективность вакцин для профилактики гриппа в группах риска (пациенты с хроническими заболеваниями, беременные). В целях контроля различных искажающих факторов и получения достоверной информации при анализе данных необходимо предусмотреть многофакторный подход.
140. С целью предотвращения заболевания гриппом новорожденных заявителям необходимо получить данные об эпидемиологической эффективности вакцин для профилактики гриппа при вакцинации матерей.
Выбор субъектов исследования
141. Для снижения потенциальной систематической ошибки отбора субъектов исследования необходимо использовать стандартизированные подходы к отбору лиц в исследованиях с активным сбором данных (например, исследования «случай - контроль с отрицательным результатом»). Необходимо фиксировать следующие сведения:
дату вакцинации и торговое наименование введенной вакцины;
данные о начале симптомов гриппоподобных заболеваний;
данные о сборе образцов;
лабораторный метод подтверждения;
подлинность штаммов гриппа, выделенных в качестве этиологических;
данные о потенциально важных искажающих факторах, таких как предшествующая вакцинация против гриппа (в течение нескольких лет, если применимо), наличие и тяжесть хронических заболеваний, анамнез курения, обращение за медицинской помощью, любая госпитализация в связи с хроническими заболеваниями за предыдущие 12 месяцев;
клинические сведения (например, госпитализация в связи с тяжелым течением гриппа).
142. В исследованиях с использованием популяционных баз данных (например, в когортных исследованиях) необходимо предусмотреть план по выявлению возможных систематических ошибок (например, включение справочных сведений, содержащихся в существующих базах данных).
Представление результатов
143. Результаты исследований эпидемиологической эффективности вакцин для профилактики гриппа необходимо представлять ежегодно по мере их получения. Анализ эпидемиологической эффективности необходимо представлять по типам и подтипам (серотипам) вируса гриппа. Результаты анализа эпидемиологической эффективности необходимо представить для каждой исследуемой популяции. В связи с тем, что оценка эпидемиологической эффективности вакцин для профилактики гриппа может проводиться в различные периоды времени, допускается представление промежуточных результатов, которые получают в режиме реального времени до получения заключительных результатов по окончанию сезонной эпидемии.
Интерпретация результатов
144. Результаты пострегистрационных исследований зарегистрированных вакцин для профилактики гриппа позволяют получить важные сведения об эффективности каждой вакцины, особенно, для новых вакцин. Несмотря на ограничения при проведении эпидемиологического надзора по оценке вакцинопрофилактики, требуется ежегодное представление данных об эпидемиологической эффективности. Отчет об эпидемиологической эффективности вакцин для профилактики гриппа включает в себя результаты исследований в течение нескольких сезонов.
145. При выявлении отклонений от ожидаемых результатов (например, снижение эффективности в связи со снижением качества отдельной вакцины или подозрением на снижение качества) требуется применение регуляторных мер.
9.6. Исследования по оценке безопасности
146. Для изучения вероятности возникновения дополнительных серьезных нежелательных явлений длительность наблюдения за вакцинируемыми должна составлять не менее 6 месяцев после введения вакцины (введения последней дозы).
147. В исследования безопасности вакцин для профилактики гриппа любого типа должны быть включены, как правило, не менее 3000 человек (общая численность популяций).
148. Заявителям необходимо согласовывать предлагаемую численность субъектов, включаемых в исследование безопасности с уполномоченными органами (экспертными организациями) государств-членов в ходе разработки программы клинического исследования, поскольку в зависимости от типа и конструкции (состава) вакцины требования могут отличаться.
149. Требования к размеру исследуемой популяции и частоте нежелательных реакций при исследовании безопасности новых вакцин для профилактики гриппа до подачи заявления о регистрации представлены в таблице.
Таблица
Необходимый размер исследуемой популяции при исследовании безопасности новых вакцин для профилактики гриппа в зависимости от их частоты и целевой группы
| № | Целевая группа (показания к применению) | Частота нежелательных реакций | Размер базы данных по безопасности для обнаружения нежелательных реакций указанной частоты |
|---|---|---|---|
| 1. | Взрослые в возрасте 18 - 65 лет или дети в возрасте от 6 месяцев до 17 лет или пожилые лица в возрасте старше 65 лет |
 1 на 1000 вакцинированных (редкие нежелательные реакции)
1 на 1000 вакцинированных (редкие нежелательные реакции)
|
достаточно базы данных из примерно 3000 субъектов исследования в единственной или в одной из указанных возрастных групп; допускается, чтобы объем данных в других возрастных группах был меньше |
| 2. | Дополнительные возрастные группы к любой из указанных в строке 1 (например, младенцы, дети, подростки, пожилые) |
 1 на 100 вакцинированных (нечастые нежелательные реакции)
1 на 100 вакцинированных (нечастые нежелательные реакции)
|
достаточно базы данных из примерно 300 субъектов исследования от каждой дополнительной возрастной группы |
| 3. | Дополнительные группы риска к любой из указанных в строках 1 и 2 (например, лица с иммунодефицитом, лица с хроническими заболеваниями) |
 1 на 100 вакцинированных (нечастые нежелательные реакции)
1 на 100 вакцинированных (нечастые нежелательные реакции)
|
достаточно базы данных из примерно 300 субъектов исследования от каждой дополнительной возрастной группы риска |
150. В каждой изучаемой возрастной группе необходимо предусмотреть соответствующую стратификацию. Например, при проведении исследований в группе детей, общая выборка целевой популяции должна составлять, по крайней мере, 3000 субъектов исследования, при этом каждая детская возрастная группа (дети грудного возраста и младенцы, дети в возрасте 2 - 8, 9 - 11 лет, подростки 12 - 14 лет и 15 - 17 лет) должна включать не менее 300 субъектов исследования, при условии отсутствия непредвиденных нежелательных реакций в детских возрастных группах. Если в исследования включают как взрослых, так и детей, общий размер популяции должен составлять 3000 взрослых плюс 300 субъектов исследования из каждой детской возрастной группы: младенцы, дети и подростки (итого приблизительно 900 детей) при условии отсутствия непредвиденных серьезных нежелательных реакций в детских возрастных группах.
151. Поскольку накоплен объем данных по безопасности по применению инактивированных безадъювантных сезонных вакцин (расщепленных и субъединичных) и моновалентных адъювантных пандемических вакцин во время беременности, достаточный для обоснования применения данных вакцин на всех этапах беременности, достоверных данных по безопасности при применении вакцин для профилактики гриппа у взрослых достаточно для обоснования использования вакцин данных типов при беременности. Для вакцин для профилактики гриппа других типов или при использовании других типов адъювантов объем данных по безопасности, который необходимо представить для обоснования их применения при беременности, требуется согласовать в рамках предрегистрационных консультаций с уполномоченными органами (экспертными организациями) государств-членов.
152. В случае если выявлены серьезные нежелательные реакции и есть опасение, что они могут быть вызваны вакциной, то потребуются дополнительные данные о безопасности.
153. Данные о безопасности, полученные при применении адъюванта отдельно от антигенов, могут служить источником дополнительных данных. Любое отклонение от требований должно быть обосновано и согласовано с уполномоченными органами (экспертными организациями) государств-членов до подачи заявления.
154. В случае применения ЖГВ в дополнение к требованиям, указанным в пунктах 148 - 153 настоящей главы, в рамках разработки программы клинических исследований необходимо представить данные по интенсивности и длительности выделения от привитых пациентов вакцинного вируса. В протоколе исследований должны быть предусмотрены меры исключения потенциального риска передачи вируса от привитых лиц к контактным лицам, особенно иммунокомпрометированным. При расширении исследований безопасности необходимо изучить воздействие ЖГВ на людей с хроническими заболеваниями, прежде всего дыхательной и сердечно-сосудистой систем.
10. Пострегистрационные требования по фармаконадзору
155. При подаче заявления о регистрации для каждой вакцины для профилактики гриппа необходимо представить план управления рисками в соответствии с требованиями, указанными в Правилах практики фармаконадзора.
156. В плане управления рисками вакцин для профилактики гриппа должны быть учтены выявленные и потенциальные риски возникновения редких и очень редких нежелательных реакций не изученных в клинических исследованиях, а также риск развития нежелательных реакций в группе иммунокомпрометированных лиц и пациентов с сопутствующими заболеваниями.
157. План управления рисками и проведение пострегистрационных исследований должны быть согласованы с уполномоченными органами (экспертными организациями) государств-членов.
10.1. Все типы вакцин для профилактики гриппа
158. План управления рисками должен предусматривать что в случае если требуется описать применение вакцины у популяций, не изученных в ходе клинических исследований (например, у иммунокомпрометированных лиц), то на пострегистрационном этапе необходимо получить соответствующие данные по иммуногенности и (или) эпидемиологической эффективности, а также необходимости применения бустерной дозы для данной популяции.
Пожилые люди и лица с хроническими заболеваниями должны составлять основную часть планируемой программы пострегистрационного мониторинга.
10.2. Сезонные вакцины для профилактики гриппа
159. В отношении сезонных вакцин для профилактики гриппа план управления рисками должен предусматривать расширенный сбор данных по безопасности вакцины. Необходимо оценить безопасность и реактогенность новых штаммов (в связи с обновлением штаммового состава сезонных вакцин) с точки зрения местных и системных нежелательных реакций при применении в различных возрастных группах, особенно у маленьких детей (при наличии показаний к применению). Такие данные необходимо получить с самого начала ежегодной кампании массовой вакцинации. Результаты необходимо своевременно представлять уполномоченным органам государств-членов.
10.3. Препандемические вакцины для профилактики гриппа
160. В отношении препандемических вакцин для профилактики гриппа план управления рисками должен предусматривать следующее:
а) проведение обсервационных исследований в случае применения препандемических вакцин для профилактики гриппа при проведении государственных программ иммунизации целевой популяции в конкретном государстве-члене;
б) при инфицировании вакцинированных лиц циркулирующими штаммами гриппа, обладающих пандемическим потенциалом (например, лица, работающие на птицефабриках, где зафиксированы случаи вспышек гриппа птиц или зафиксированы случаи инфекции человека вследствие контактов с больными животными), необходимо осуществлять сбор сведений о всех случаях возникновения заболевания у вакцинированных. Сбор дополнительных данных следует осуществлять в популяциях, которые не изучались в рамках клинических исследований;
в) мониторинг эпидемиологической эффективности вакцины, применяемой в условиях пандемии. Такие данные будут информативными для планирования стратегий будущих препандемических вакцинаций. Если данные становятся доступными достаточно рано до признания пандемии и они свидетельствуют о протективных свойствах вакцины, это позволит использовать все доступные пандемические вакцины для вакцинации преимущественно ранее невакцинированных популяций.
10.4. Пандемические вакцины для профилактики гриппа
161. В случае применения пандемических вакцин план управления рисками должен предусматривать следующее:
а) оценку эпидемиологической эффективности и безопасности применения пандемических вакцин у беременных, поскольку в условиях пандемии именно они будут являться первой целевой популяцией при проведении кампании по вакцинации. Вакцинация беременных женщин может дополнительно защитить новорожденных от инфекции. Эффективность и безопасность должны контролироваться, и такие исследования необходимо планировать во время процедуры регистрации;
б) сбор данных об иммуногенности, эпидемиологической эффективности и безопасности применения вакцин для профилактики гриппа во время пандемии, должен осуществляться совместными усилиями фармацевтических компаний и органов системы здравоохранения. Своевременный обмен и анализ полученных данных позволят быстро реагировать на необходимость внесения изменений в штаммовый состав пандемической вакцины, режим и программу проведения вакцинации в ходе пандемии;
в) в дополнение к оценке частоты местных и системных нежелательных реакций в ранний поствакцинальный период, необходимо предусмотреть потенциальные риски возникновения специфических долгосрочных, а также редких и очень редких нежелательных реакций, (например, риск нарколепсии или синдрома Гийена-Барре). В отношении пандемических вакцин необходимо получить масштабные данные о безопасности в рамках полевого применения;
г) перед подачей заявления на изменение пандемического штамма держатели регистрационного удостоверения должны обсудить и согласовать с уполномоченными органами (экспертными организациями) государств-членов планы по расширенному мониторингу безопасности, подлежащему проведению в пандемический период.
11. Общая характеристика лекарственного препарата и маркировка вакцин для профилактики гриппа
162. Не имеется типовой общей характеристики лекарственного препарата и типовой маркировки для отдельных типов вакцин для профилактики гриппа. Общую характеристику лекарственного препарата и маркировку лекарственного препарата заявителю необходимо формировать, основываясь на собственных данных, полученных для каждого типа вакцины.
Глава 30. Указания по разработке вакцин против оспы (осповакцин)
1. Общие положения
1. В 1980 г. Всемирной организацией здравоохранения (ВОЗ) была принята резолюция о достижении полной ликвидации натуральной оспы, путем применения вакцины на основе вируса из семейства ортопоксвирусов, отличного от вируса натуральной оспы, но обеспечивающего кросс-протективность против вируса натуральной оспы. В настоящее время осповакцина на случай чрезвычайной ситуации, а также для вакцинации лиц группы риска, производится по технологии существовавшей в период глобальной ликвидации оспы (то есть до 1980 г.) и не в полной мере соответствует объему требований, установленных к производству биологических препаратов и надзору за их безопасностью.
2. Вакцины против оспы делятся на три группы:
а) вакцины первого поколения - большинство выпускаемых до 1980 года осповакцин, произведенных с использованием живых животных (телят, овец, буйволов и кроликов). Кролики, в частности, использовались для наработки посевного материала, а основными видами животных для производства вакцины были телята и овцы. Сохранение производства с использованием живых животных имеет свои преимущества, поскольку ранее было доказано, что полученная вакцина обладает достаточной эффективностью, а все процессы производства хорошо задокументированы и могут быть развернуты в короткие сроки. Однако такие вакцины не всегда позволяют обеспечить выполнение требований к процессу производства и качеству полученного продукта в части микробиологического загрязнения;
б) вакцины второго поколения, использующие системы культур тканей (почек кролика и крупного рогатого скота) или куриные эмбрионы. Вакцины на основе вируса осповакцины, приготовленные на куриных эмбрионах имеют опыт применения в странах Южной Америки, Израиле. Опыт промышленного производства осповакцин на культуре клеток ограничен (в Японии была произведена вакцина на куриных эмбриональных фибробластах (chick embryo fibroblasts, CEFs), которая имела благоприятный профиль безопасности, но данные по ее эффективности были недостаточно задокументированы). Они содержат реплицирующийся вирус и вводятся в организм методом скарификации кожи;
в) вакцины третьего поколения, содержащие вирусы, произведенные при помощи генной инженерии и (или) технологий переноса генов.
3. В настоящее время основным видом вакцин против оспы являются вакцины второго поколения. С учетом того, что вирус осповакцины довольно широко используется в качестве вектора для экспрессии антигенов (особенно в исследованиях, связанных с вирусом иммунодефицита человека (ВИЧ)), имеется значительный опыт производства небольших его серий, подходящих для клинического применения с использованием современных методик, главным образом, CEFs, диплоидных клеток человека MRC-5 и линии клеток почки африканской зеленой мартышки, Vero. Однако в то время как иммунный ответ на переносимые гены тщательно изучался, ответ на сам вирус осповакцины документировался гораздо менее детально, а опыт изучения эффективности собственно вакцин против оспы, выращенных на культуре клеток, отсутствует или ограничен. Уровень качества в отношении микробиологической чистоты, который можно достигнуть для данных вакцин, гораздо выше такого уровня для вакцин, произведенных с использованием живых животных. Однако реальный уровень безопасности и эффективности вакцины будет зависеть не только от отсутствия в ней примесей, но также и от качества вирусного посевного материала. Несмотря на высокую микробиологическую чистоту, следует отметить, что вакцины, выращенные на тканевой культуре, могут быть связаны с такими же серьезными нежелательными реакциями, что были выявлены у вакцин животного происхождения, наиболее широко использовавшихся в программе ликвидации оспы. Следует отметить, что производство на культуре тканей предполагает адаптацию вирусного штамма к используемому клеточному субстрату и эффект от подобной адаптации в отношении иммуногенности и безопасности вакцины подлежит изучению, а свойства произведенной вакцины устанавливаются на соответствующей животной модели.
2. Сфера применения
4. В свете потенциальной возможности использования вируса натуральной оспы как фактора биотерроризма, а также возникновения мутаций в семействе ортопоксвирусов у вируса оспы обезьян настоящая глава содержит указания по разработке вакцин второго поколения против инфекций, вызываемых вирусами оспы, потенциально патогенными для человека, связанные с производством, программами доклинической и клинической разработки противооспенных вакцин, произведенных с использованием вируса осповакцины, наработанного на культуре тканей или куриных эмбрионах. Системы культур тканей включают в себя первичные культуры клеток, диплоидные или перевиваемые клеточные линии.
5. Настоящая глава содержит указания по проведению оценки уполномоченными органами (экспертными организациями) государств-членов качества, безопасности и иммуногенности вакцин против оспы второго поколения, произведенных с применением методик, описанных в пункте 4 настоящей главы. В настоящей главе приводятся указания по представлению заявителем данных, необходимых для получения регистрационного удостоверения на вакцину против оспы.
6. Положения настоящей главы не применяются в отношении вакцин против оспы, произведенных на основе вируса осповакцины, выращенного с использованием живых животных (вакцины против оспы первого поколения). В отношении требований к вакцинам против оспы первого поколения производители вправе применять указания Всемирной организации здравоохранения в отношении организации производства и контроля качества вакцин против оспы. Вакцины против оспы третьего поколения также исключены из сферы применения настоящей главы, однако приведенные в ней положения допускается применять при производстве и оценке таких вакцин.
7. В отношении производства, контроля качества и клинических исследований вакцин против оспы второго поколения применяются требования настоящих Правил, Фармакопеи Союза (а при отсутствии в ней - фармакопей государств-членов) в отношении:
использования клеточных субстратов;
контроля посторонних биологических агентов;
использования стад кур в производстве вакцин;
использования бычьей сыворотки.
3. Выбор вакцинного штамма
3.1. История получения штамма
8. Для целей выполнения требований настоящей главы при производстве вакцин допускается применение следующих штаммов вируса осповакцины (генетическая связь данных штаммов друг с другом остается неизученной), которые использовались в период ликвидации оспы:
а) штамм Lister, разработанный в институте Листера (синоним «штамм Elstree» или «Lister/Elstree», поскольку указанный институт располагался в городе Элстри в Великобритании). Плавный посевной материал был получен Национальным институтом здравоохранения Нидерландов в сотрудничестве с ВОЗ и хранится там до настоящего времени. Он был приготовлен путем двух пассажей на лимфе телят из оригинального материала, полученного в 1961 г. на лимфе овец. Главный посевной материал был отправлен в центры в Париже, Токио, Атланте и Москве. Материал также был предоставлен производителям вакцины по всему миру, при этом многие из них независимо получили посевной материал из своей производственной культуры. Использовался 23 из 59 производителей вакцины во время эрадикационной кампании;
б) штамм NYCBOH (New York City Board of Health (Совет no вопросам здравоохранения города Нью-Йорк)), который использовался в Северной и Южной Америке, а также в Западной Африке. Семь производителей вакцины использовали этот штамм. Штамм ЕМ-63, использовавшийся в СССР, являлся производным от штамма NYCBOH и широко применялся во время кампании по искоренению оспы в Индии;
в) другие штаммы вируса осповакцины:
штамм Paris, использовался 7 производителями вакцины на четырех континентах;
штаммы Copenhagen, Bern и Temple of Heaven (последний - использовался в Китае и, возможно, был более иммуногенным по сравнению с другими штаммами).
9. Не допускается без соответствующего обоснования использование при производстве вакцин нереплицирующихся штаммов, таких как:
штамм модифицированного вируса Ankara (MVA), представляющий собой экспериментальный штамм, созданный путем многократных пассажей на куриных эмбриональных фибробластах (CEFs), в результате чего является высокоаттенуированным вирусным штаммом, способным к ограниченному размножению в культуре человеческих клеток. Штамм применялся в Германии для иммунизации нескольких сотен тысяч людей в качестве первичной вакцинации для смягчения нежелательных явлений. Эпидемиологическая эффективность данного штамма неизвестна. Аттенуированные штаммы допускается включать в изучение и разработку с целью возможного применения у иммунокомпрометированных лиц;
прочие нереплицирующиеся штаммы, например, NYVAC.
3.2. Доступность штамма
10. Производителям необходимо оценить доступность штамма для организации производства на регулярной основе.
Главный посевной материал штамма Lister хранится в Национальном институте здравоохранения Нидерландов. Запрос на получение должен первоначально должен быть направлен в ВОЗ.
Запросы на получение штамма NYCBOH выполняются в Центр по контролю и профилактике заболеваний (Centers for Disease Control and Prevention, CDC) в США.
Информация о доступности и возможности получения других штаммов должна уточняться производителем самостоятельно.
3.3. Факторы, влияющие на выбор штамма
11. Профили безопасности различных штаммов могут варьироваться, а имеющиеся сравнительные данные крайне ограничены. Более задокументирована информация по вакциноассоциированным побочным реакциям в отношении штаммов Lister и NYCBOH в сравнении с другими штаммами, использовавшимися во время осуществления программы по ликвидации оспы.
12. В связи с тем, что производство клеточной культуры и клональная селекция могут привести к изменению характеристик вируса, представляется затруднительным дать четкие указания по выбору штамма. Тем не менее при этом учитываются следующие факторы:
профиль безопасности и биологические характеристики родительского штамма;
предполагаемая эффективность (основанная на успешной ликвидации оспы) вакцины, изготовленной на основе родительского штамма, оцениваемая по доле успешно вакцинированных и применению в полевых условиях;
история родительского штамма;
ростовые характеристики выбранного штамма в клеточном субстрате;
имеющийся у производителя опыт изготовления штамма для противооспенных вакцин первого поколения.
3.4. Стандартные образцы
13. Международный стандартный образец (произведенный с использованием пашины овцы), изготовленный на основе штамма Lister, доступен в Национальном институте биологических стандартов и контроля Великобритании (National Institute for Biological Standards and Control, NIBSC). Данный международный стандартный образец предоставляется в небольшом количестве и предназначен для оценки специфической активности стандартных образцов предприятия. Допускается использование для этих же целей образцов NIBSC, эквивалентных международному стандартному образцу для противооспенной сыворотки.
14. Допускается использовать подходящие национальные стандартные образцы, откалиброванные по международному стандартному образцу и произведенные другими контрольными лабораториями государства-члена с целью калибровки стандартных образцов производителя. Принимая во внимание сокращение запасов международных стандартных образцов, в перспективе допускается их замена новыми наработанными стандартными образцами при их надлежащей аттестации. Потенциальный производитель вакцины против оспы обязан предоставить информацию о направленных им запросах относительно о доступности новых стандартных образцов.
4. Требования к качеству вакцин против оспы
4.1. Посевные культуры вакцинного штамма
Приготовление культуры вакцинного штамма
15. Должна использоваться система посевной культуры вакцинного штамма, включающая в себя главную посевную культуру и рабочую посевную культуру. Разработка и создание данных культур, их условия хранения и срок годности должны быть подробно описаны. Культуру готовят пассажем выбранного вакцинного штамма в клеточном субстрате, который будет использоваться в производстве вакцины, или in ovo, если планируется данный метод производства вакцины. Общепризнано, что продолжительный многократный пассаж вирусного штамма в культуре тканей или in ovo может снизить его иммуногенные свойства, в связи с чем количество пассажей от родительского штамма вируса к главной и рабочим посевным культурам должно быть ограничено и должным образом обосновано. При производстве конечной серии вакцины, количество пассажей вакцинного вируса из рабочей посевной культуры не должно превышать количества пассажей при производстве вакцины для клинических исследований, если иное не будет соответствующим образом обосновано и одобрено уполномоченным органом (экспертной организацией) государства-члена. Вакцины должны производиться из рабочей посевной культуры с минимальным количеством промежуточных пассажей.
16. Приготовление главной посевной культуры из штамма родительского вируса может включать клонирование при помощи методики бляшкообразования или другие формы клональной селекции. Клонирование при помощи методики бляшкообразования может быть полезным в удалении возможных побочных вирусов, находящихся в вакцине наряду с родительским штаммом, а также делает популяцию вакцинного штамма более однородной. При этом необходимо учитывать, что в результате может быть отобрана атипичная субпопуляция, в связи с этим необходимо будет описать характеристики главной посевной культуры в различных системах, в том числе в условиях in vivo.
Установление характеристик главной посевной культуры
17. Необходимо сделать максимально полное описание главной посевной культуры. В данное описание необходимо включить полную задокументированную историю родительского штамма, полное описание характеристик в сравнении со штаммом родительского вируса, хотя для данной цели доступно только ограниченное количество родительских штаммов. Установление характеристик должно включать следующие пункты (отсутствие любого пункта должно быть обосновано):
антигенный анализ с использованием специфичной антисыворотки и (или) моноклональных антител;
биологические исследования: определение титров инфекционности, культивирование на хориоаллантоисной мембране с образованием местных вирусных поражений, выход вирусного материала in vitro и ростовые характеристики in vivo на подходящей животной модели;
генетические анализы, такие как: картирование с использованием разных рестрикционных ферментов и Саузерн-блоттинга, ПЦР-анализ и ограниченные исследования методом секвенирования;
стабильность генетических и фенотипических характеристик после пассажа в субстрат, используемый для производства;
исследование нейровирулентности (в соответствии с разделом 5 настоящей главы);
исследования иммуногенности (в соответствии с разделом 5 настоящей главы).
Количество исследований зависит от предшествующей истории оригинального штамма, истории клеток, использовавшихся в его приготовлении, применения клональной селекции и от природы всех реагентов, использовавшихся в приготовлении посевной культуры.
Необходимо определить вирусные титры главной и рабочей посевной культуры, а также проводить мониторинг вирусного титра рабочей культуры во время хранения с целью обеспечения однородности вакцинной продукции.
Исследование на наличие посторонних агентов
18. При проведении исследований на наличие посторонних биологических агентов необходимо принимать во внимание историю пассажей родительского вирусного штамма и клеточных субстратов, использовавшихся в производстве клеточных культур. Исторически, в XIX веке, для размножения вируса использовались овцы, козы, телята, буйволы, кролики и люди. Однако вряд ли можно установить полную историю пассажей вирусного штамма, поэтому сохраняется возможность того, что любой штамм мог пересеваться в пределах более чем одного вида. Например, посевной материал ВОЗ штамма Lister, был получен на лимфе телят из материала, первоначально полученного на лимфе овцы.
19. Основным методом исследования штамма живой вирусной вакцины на наличие посторонних вирусов являются испытания in vitro и in vivo с предварительной нейтрализацией вакцинного вируса. Испытания вирусного сбора в клеточной культуре, полученного с использованием куриных эмбрионов, культуры первично трипсинизированных клеток, необходимо проводить на трех типах клеток:
гомологичной, приготовленной из другой партии производственного субстрата (например, фибробласты куриных или перепелиных эмбрионов);
клетках человека или обезьяны (например, диплоидные клеточные культуры человека Л-68 или MRC-5, или клетки Vero);
наиболее чувствительных клетках для выявления вирусов - возможных контаминантов используемого субстрата (например, почки эмбриона кур).
Если вирус выращен на клетках обезьяны или человека в системе банков клеток, то нейтрализованный вирусный сбор испытывают на отдельной культуре этих клеток. Вирус осповакцины крайне трудно нейтрализовать до той степени, которая необходима в ходе проведения данных испытаний. Необходимо учитывать, что вирусный материал может содержать фенотипически различные внутриклеточные и внеклеточные формы, при этом первые нейтрализуются легче, чем последние. Необходимо отметить, что если исследуемый вакцинный вирус перед нейтрализацией был разведен до более низкого титра с целью достижения полной нейтрализации, это может негативно повлиять на чувствительность при обнаружении посторонних вирусов. Если требуемые испытания нельзя провести из-за невозможности полностью нейтрализовать вирус, посевной материал можно развести до концентрации, соответствующей разведению, используемому для инокулята в производстве вакцины до испытания на посторонние вирусы.
20. Основное исследование должно проводиться в соответствии с требованиями Фармакопеи Союза. При отсутствии соответствующей статьи в Фармакопее Союза исследование может проводиться согласно требованиям фармакопеи референтного государства. Также могут потребоваться дополнительные исследования, которые могут включать в себя методы амплификации нуклеиновых кислот для определенных вирусов, а также испытания на ретровирусы с использованием обратной транскриптазы. В случае если полная нейтрализация вакцинного вируса не может быть достигнута, допускается использовать альтернативные методы обнаружения посторонних специфических загрязнений. При исследовании на наличие определенных вирусов, необходимо также обращать внимание на восприимчивость к ним произведенного клеточного субстрата (в соответствии с подразделами 4.2 - 4.4 настоящей главы).
21. Если в разработке или производстве главной и рабочей посевной культур использовались материалы крупного рогатого скота или иных видов животных, источников губчатой энцефалопатии, необходимо учитывать требования актов органов Союза по минимизации риска передачи агентов губчатой энцефалопатии животных посредством лекарственных препаратов для медицинского применения.
22. Испытания на стерильность, содержание микобактерий и микоплазм должны проводиться в соответствии с требованиями Фармакопеи Союза, а при отсутствии в ней - с требованиями фармакопей государств-членов. При приготовлении главного посевного материала необходимо применять процедуры, которые способствуют удалению из материала посторонних агентов. Поскольку удаление или инактивация данных агентов на любом этапе производственного процесса живой вакцины против оспы практически невозможно, наличие посторонних агентов в посевных культурах не допускается.
4.2. Банк клеток
23. Система банков клеток должна состоять из главного банка клеток и рабочего банка клеток. Получение и установление характеристик банков клеток необходимо осуществлять в соответствии с настоящими Правилами и требованиями Фармакопеи Союза, а при отсутствии в ней - с требованиями фармакопей государств-членов.
24. Необходимо исследовать потенциальную вероятность инфицирования клеточного субстрата патогенными агентами, которые могут присутствовать в вирусном посевном материале. Так как данная вероятность имеет большую зависимость от истории родительского вакцинного штамма, производителю необходимо выполнить условия, указанные в подразделе 4.1 настоящей главы.
25. Клеточная линия, используемая для приготовления живых вакцин, не должна обладать туморогенными свойствами на любом уровне удвоения популяции, используемой для производства вакцины.
4.3. Первичные клеточные культуры
26. Так как первичные клеточные культуры не используются для создания банка клеток, невозможно провести их всестороннюю проверку и установление характеристик, как это делается в отношении диплоидных или перевиваемых клеточных линий.
27. Требования, указанные в подразделе 4.2 настоящей главы, относятся к клеточным культурам, используемым для создания банка клеток и дополняют требования по использованию первичных культур приложения к главе 1 настоящих Правил.
28. Первостепенную важность при использовании первичных клеточных культур имеет недопущение их загрязнения посторонними агентами. Животные, используемые в качестве источника ткани для приготовления первичного клеточного субстрата, должны быть выращены в закрытых, беспатогенных (specified pathogen free - SPF) колониях или стадах и использоваться исключительно с целью получения первичных клеточных культур. Данные колонии или стада должны строго и регулярно контролироваться на предмет наличия и поддержания беспатогенного (SPF) статуса. Стада кур, используемых в качестве источника первичных клеточных культур, должны соответствовать требованиям Фармакопеи Союза, а при отсутствии в ней - требованиям фармакопей государств-членов.
29. Необходимо исследовать потенциальную вероятность инфицирования клеточного субстрата патогенными агентами, которые могут присутствовать в вирусном посевном материале. Так как данная вероятность зависит от истории родительского вакцинного штамма, производителю также учитывать положения подраздела 4.1 настоящей главы, а также данные, полученные ранее при организации промышленного производства и использования коревой и паротитной живых аттенуированных вакцин, произведенных на первичных культурах куриных эмбриональных фибробластов, приведенные в указаниях ВОЗ для живых моновалентных и комбинированных вакцин против кори, паротита и краснухи (в частности данных указаний, касающихся получения клеточных культур из эмбрионов птиц).
4.4. Куриные эмбрионы
30. Ключевой особенностью использования куриных эмбрионов является недопущение их загрязнения посторонними агентами. В связи с этим используемые яйца должны быть получены от закрытых, беспатогенных (SPF) здоровых стад, которые содержатся исключительно для целей получения посевного материала или производства вакцины. Данные стада, в соответствии с требованиями Фармакопеи Союза, должны строго и регулярно контролироваться на предмет наличия и поддержания беспатогенного (SPF) статуса. Для производства живой аттенуированной вакцины против оспы должны использоваться яйца кур только из стад, прошедших указанный контроль.
31. Необходимо исследовать потенциальную вероятность инфицирования куриных эмбрионов патогенными агентами, которые могут присутствовать в вирусном посевном материале. Так как данная вероятность зависит от истории родительского вакцинного штамма, производителю также необходимо обратиться к подразделу 4.1 настоящей главы.
4.5. Производство вакцины
32. В целом, производство вакцин против оспы второго поколения аналогично производству других живых вирусных вакцин, включая технологические стадии, в связи с чем основные требования к производству и контролю за данными вакцинами против оспы будут практически идентичны требованиям к производству других живых вакцин.
33. Заявителю необходимо привести полное описание процесса производства вакцины. Клетки, полученные из банков клеток, первичные клеточные культуры млекопитающих или птиц и (или) эмбрионы птиц должны соответствовать требованиям настоящих Правил и Фармакопеи Союза, а при отсутствии в ней - фармакопей государств-членов. Все материалы, используемые в процессе производства, должны быть должным образом описаны и быть соответствующего качества. Одобренные сыворотки животного происхождения используются только при условии того, что уровень остаточных загрязнений в них был снижен до допустимых пределов. При использовании бычьей сыворотки необходимо руководствоваться требованиями Фармакопеи Союза по минимизации риска передачи губчатой энцефалопатии животных. При использовании любых материалов животного происхождения требуется соблюдение требований актов органов Союза в сфере ветеринарного законодательства, обращения лекарственных средств для медицинского применения и соответствующих статей Фармакопеи Союза, а при отсутствии в ней - соответствующих статей фармакопей государств-членов. Пенициллин, другие бета-лактамные антибиотики и стрептомицин не должны использоваться в процессе производства вакцины, и (или) в качестве добавки к конечному продукту.
Культивирование вакцинного вируса
34. Клеточную суспензию в концентрации, используемой для посева клеточных культур-продуцентов вакцины (не менее 5% или не менее 500 мл, если этот объем будет меньшим) не подвергают заражению вирусом и исследуют в качестве контрольных клеток производственной клеточной культуры. Если вакцина производится с использованием банка клеток, контрольные клетки должны выдерживать испытания на обнаружение цитопатических изменений и феномена гемадсорбции. Контрольные клетки изучают под микроскопом в течение периода инкубации производственной клеточной культуры до времени сбора вируса с культуры, но не менее 14 дней после посева, на отсутствие любой цитопатической реакции вирусного происхождения. Результаты испытания считают достоверными, если не менее 80% контрольных клеток остаются жизнеспособными. Не менее 25% контрольных клеток, не ранее, чем на 14-й день после посева, испытывают на наличие феномена гемадсорбции путем добавления эритроцитов морских свинок.
35. Если вакцина производится с использованием первичных клеточных культур контрольные клетки, отобранные в день инокуляции производственной серии, подвергают испытанию, описанному в подразделах 4.1 и 4.3 настоящей главы.
36. Если при исследовании контрольных клеток в одном из испытаний обнаружены цитопатические изменения или феномен гемадсорбции, то вирусный сбор, полученный в производственной культуре, не должен быть использован для приготовления вакцины.
Однократные сборы вируса
37. Необходимо описать выбранный способ сбора вакцинного продукта. Вирус осповакцины представлен как во внутриклеточной, так и во внеклеточной формах. Протокол сбора вируса должен учитывать то, что внутриклеточную форму вируса необходимо будет высвободить из производственной клеточной культуры или куриного эмбриона. Обычно однократные сборы вируса объединяются в общий пул, из которого готовится конечный балк. Однако, в зависимости от размера единицы продукции, одна или несколько из указанных стадий могут не потребоваться.
38. Необходимо провести испытания на подлинность в отношении собранного вируса. Необходимо определить вирусный титр после фильтрации или стадии очистки, либо с помощью теста на хориоаллантоисной мембране (результат выражают в единицах сформировавшихся оспин на мл) либо при помощи валидированной методики титрования вируса в культуре клеток, при этом результаты выражают в CCID50 (50% инфекционная доза для клеточной культуры) или в единицах бляшкообразования (каждая бляшка принимается за одну инфекционную единицу). Для валидации методики титрования необходимо включать в исследование референтный препарат. Необходимо установить минимально допустимое значение титра для использования однократного сбора вируса в приготовлении вирусного пула или конечного балка.
39. Исследования на посторонние агенты необходимо проводить на каждом однократном сборе согласно требованиям Фармакопеи Союза, а при отсутствии в ней - требованиям фармакопей государств-членов, при этом дизайн исследований должен учитывать трудность нейтрализации вируса осповакцины и способность вируса осповакцины вызывать в культуре цитопатогенное и гемадсорбирующее действие. Испытуемый материал после предварительной нейтрализации вируса иммунной сывороткой или удаления вируса исследуют в культуре клеток на наличие цитопатических изменений и феномена гемадсорбции, как описано в подразделах 4.1 настоящей главы и в настоящем подразделе. В случае если полная нейтрализация вируса не достигается, испытания в культуре клеток заменяются специфическими испытаниями, например, с помощью метода амплификации нуклеиновых кислот и иммунохимическими методами. При этом тест-системы, способные выявить возможные посторонние агенты, должны быть нечувствительны к вирусу осповакцины.
40. Способ производства (а именно: использование клеточной культуры из банка клеток, первичной клеточной культуры или куриных эмбрионов) должен учитывать природу каждого вируса, подвергаемого исследованию. Все тест-системы должны быть надлежащим образом валидированы, а предел обнаружения для потенциальных патогенных агентов необходимо документально зафиксировать и обосновать.
41. Необходимо оценить безопасность контрольных клеток относительно потенциальной контаминации посторонними агентами при помощи микроскопического исследования для обнаружения цитопатических эффектов, а также при помощи других испытаний в соответствии с Фармакопеей Союза, а при отсутствии в ней - в соответствии с фармакопеями государств-членов. Для производства вакцины против оспы с использованием куриных эмбрионов необходимо оценивать контрольные куриные эмбрионы из каждой серии, используемой для производства, на наличие гемагглютинирующих агентов и вирусов лейкоза птиц.
42. Также необходимо проводить испытания однократных сборов вируса на стерильность и содержание микоплазм согласно требованиям Фармакопеи Союза, а при отсутствии в ней - требованиям фармакопей государств-членов.
Вирусные пулы
43. Необходимо разработать и описать стратегию объединения однократных сборов вируса в вирусный пул. При этом необходимо использовать только однократные сборы вируса, успешно прошедшие испытания, описанные в пунктах 37 - 42 настоящей главы. Вирусный пул должен являться частью стадии очистки и должен быть подвергнут концентрированию с целью получения необходимого вирусного титра. Все технологические операции с вирусным пулом должны быть детально описаны. Необходимо проводить испытания вирусных пулов на стерильность согласно Фармакопее Союза.
Конечный балк
44. Конечный балк допускается приготавливать из одного или нескольких вирусных пулов, а также получать из однократного сбора вируса. При этом необходимо использовать только однократные сборы вируса и вирусные пулы, успешно прошедшие испытания, описанные в пунктах 37 - 43 настоящей главы.
Состав
45. При изготовлении конечного балка необходимо учитывать следующие аспекты. Необходимо подтвердить, что все вещества, добавляемые к продукту во время приготовления конечного балка (растворители, стабилизаторы или любые другие наполнители) не ухудшают эффективность и безопасность вакцины в используемых концентрациях. Для долгосрочного хранения необходимо использовать лиофилизацию. При восстановлении лиофилизированной вакцины глицерин в разбавителе служит для стабилизации вакцины и способствует ее лучшему прикреплению к поверхности бифуркационной иглы или ланцета для вакцинации. При этом использование бифуркационной иглы для введения вакцины является предпочтительным. В восстановительную среду возможно добавление красителя, если это не ухудшает безопасность, активность и стабильность после вскрытия и в ходе применения восстановленной вакцины.
46. Добавление антибиотиков в качестве консервантов не допускается. Для многодозовых составов следует оценить необходимость применения эффективных антимикробных консервантов, с учетом возможной микробной контаминации во время использования и рекомендованного максимального срока годности (срока хранения) после вскрытия упаковки и восстановления вакцины. В случае применения антимикробного консерванта, он не должен ухудшать показатели качества и безопасности вакцины.
Испытания конечного балка
47. Титр конечного балка должен учитывать его возможное снижение во время процессов наполнения, лиофилизации и на протяжении всего срока годности (срока хранения) вакцины. Качественный и количественный анализ конечного балка вакцины должен включать в себя испытания по оценке содержания общего белка, испытания вспомогательных веществ, контроль остаточных сывороточных белков животного происхождения (например, бычьего сывороточного альбумина). Полученные результаты должны соответствовать результатам, указанным в спецификациях.
48. В зависимости от типа используемого субстрата (пересеваемых клеточных линий) следует разработать соответствующие и должным образом валидированные испытания на остаточные ДНК и белки клетки-хозяина. Каждая порция конечного балка должна быть подвергнута испытанию на стерильность согласно Фармакопее Союза.
49. Конечный вирусный балк необходимо исследовать на нейровирулентность на подходящей животной модели (например, интрацеребральная инокуляция на мышах) с целью подтверждения соответствия препарата его биологическому фенотипу. Исследование нейровирулентности также может быть проведено на этапе вирусного пула. В исследование можно включить референтный препарат, которым может быть предыдущая порция конечного балка или вирусного пула. По мере накопления данных об использовании препарата, необходимость проведения данного исследования может быть пересмотрена.
50. До фасовки в первичную упаковку (наполнение контейнеров) конечный балк необходимо хранить при условиях, которые продемонстрировали способность сохранять его вирусную активность.
Наполнение продукции
51. Наполнение и маркировка контейнеров должны осуществляться согласно требованиям к производству биологических лекарственных препаратов в соответствии с Правилами производственной практики.
4.6. Контрольные испытания конечного продукта вакцины
52. После восстановления образцы контейнеров из каждой серии конечного вакцинного продукта должны быть подвергнуты испытаниям на стерильность, подлинность и активность. Для вакцин, приготовленных с использованием куриных эмбрионов, содержание эндотоксинов должно соответствовать критериям приемлемости, определенным на основе результатов анализов производственной серии.
Исследования активности
53. ВОЗ установлен титр для первого поколения противооспенных вакцин, составляющий не менее 1x108 единиц сформировавшихся оспин на мл. Данное значение должно служить основой для установления минимального титра для вакцин второго поколения. Титр конечного продукта, установленный в спецификации на выпуск, должен быть надлежащим образом подтвержден, основываясь на доклинических и (или) клинических данных, полученных для разрабатываемой вакцины.
54. Для определения активности необходимо применять методики исследования на хориоаллантоисной мембране (результат выражается в единицах сформировавшихся оспин на мл) либо при помощи валидированной методики титрования вируса в культуре клеток, при этом результаты выражаются в CCID50 (50% инфекционная доза для клеточной культуры) или в единицах бляшкообразования (каждая бляшка принимается за одну инфекционную единицу). Для валидации исследования в него необходимо включать референтный препарат.
Аномальная токсичность
55. Необходимо провести общее исследование по оценке безопасности на отсутствие аномальной токсичности, связанной с конечным продуктом. Данное исследование осуществляется в соответствии с Фармакопеей Союза только на протяжении периода валидации процесса производства.
При необходимости производитель вправе использовать результаты испытания на аномальную токсичность последовательных промышленных серий для подтверждения постоянства производства.
4.7. Стабильность
56. Необходимо определить и надлежащим образом обосновать спецификации на выпуск и конец срока годности (срока хранения). Необходимо подтвердить сохранение активности вакцины на протяжении всего периода валидации процесса производства. Любое снижение активности при хранении должно быть надлежащим образом оценено. Значительное снижение активности (даже не выходящее за допустимые пределы), свидетельствует о нестабильности вакцинного продукта. Для лиофилизированных вакцин также необходимо разработать надлежащие критерии приемлемости при исследовании термостабильности (например, 4 недели при 37°С).
5. Доклинические исследования
5.1. Основные положения
57. Фармакологические и токсикологические характеристики вакцины-кандидата необходимо оценить в исследовании с референтной вакциной, представляющей собой оригинальный вакцинный штамм, из которого была произведена вакцина. Данный оригинальный штамм должен быть произведен в соответствии с Указаниями ВОЗ в отношении производства и контроля качества вакцин против оспы 2003 г.
58. Исследование вируса натуральной оспы на животных моделях довольно затруднительно. В то же время вирус осповакцины обладает кросспротективностью в отношении вируса натуральной оспы и других ортопоксвирусов, патогенных для млекопитающих. В отношении выбора релевантного вида животного для исследования известно, что вирус осповакцины способен вызывать должный иммунный ответ у нескольких видов (мыши, кролики, обезьяны).
59. Исследования токсичности требуется проводить в отношении главной посевной культуры, рабочих посевных культур и конечного продукта. Проведение фармакодинамических исследований необходимо только для конечного продукта.
60. Для исследования на животных необходимо выбирать актуальные модели, описанные в современной научной медицинской литературе. Использование животных моделей должно быть подробно описано и обосновано с учетом актуальных разработок и исследований в данной области. Заявителям необходимо использовать указанные модели в исследовательских учреждениях, на базе которых они уже развернуты (введены в рабочий процесс), чтобы избежать затратного по ресурсам и времени процесса внедрения сложных моделей в работу исследовательского учреждения, не имеющего подобного опыта.
5.2. Фармакодинамика
61. Оценка протективного эффекта вакцин против оспы не может проводиться на людях ни в исследованиях эффективности, ни в исследовании методом экспериментального инфицирования привитых добровольцев. В связи с этим оценка потенциального протективного эффекта должна до определенной степени основываться на результатах соответствующих исследований на животных. Проведение данных исследований является обязательным до начала планирования любых клинических исследований.
Первичная фармакодинамика
62. Первичной конечной точкой в исследованиях на животных должна быть протективная эффективность вакцины-кандидата в сравнении с оригинальной вакциной при экспериментальном инфицировании патогенным ортопоксвирусом.
63. Доклинические исследования вакцин против оспы второго поколения, даже в релевантных животных моделях, могут заменить клинические исследования у человека только частично. Любая используемая животная модель должна быть максимально, насколько это возможно, близкой к человеческому организму. Кросспротективность должна быть показана к двум различным патогенным ортопоксвирусам на двух различных видах млекопитающих. При этом необходимо применять поэтапный подход, не используя приматов в животных моделях на ранних стадиях доклинической и фармацевтической разработки. Изучение протективной активности конечного продукта на мышах является необходимым до начала планирования клинических исследований. Окончательное подтверждение протективной эффективности конечного продукта должно быть получено в исследовании на обезьянах, при этом данное исследование может проводиться параллельно с первыми клиническими исследованиями у человека.
64. В качестве модели без использования приматов могут выступать мыши линии BALB/c (данная модель детально писана в научной медицинской литературе). Первичной конечной точкой будет являться протективность в отношении летальной респираторной инфицирующей дозы ортопоксвируса, например, вируса коровьей оспы. Вирусиндуцированные симптомы (например, снижение веса тела) при этом будут являться вторичными конечными точками. Полученные результаты должны быть, по меньшей мере, эквивалентны результатам, полученным для препарата сравнения. Дополнительные данные по протективной активности допускается получать в исследованиях подбора доз и при использовании вирусов с различной вирулентностью. Для введения вакцины рекомендуется внутрикожное введение, предпочтительно скарификация.
65. В научной медицинской литературе использование обезьян в качестве модели подробно описано для макак-крабоедов с применением вируса обезьяньей оспы в виде аэрозоля. Первичной конечной точкой является протективность в отношении летальной дозы. Проводить такие исследования необходимо в лабораториях с уровнем биологической безопасности BSL-3.
66. При использовании моделей мышей линии BALB/c и макак-крабоедов необходимо получить следующие дополнительные данные:
индукция образования пустулы или рубца;
индукция образования антител и клеточно-опосредованного иммунного ответа. Для анализа образования антител допускается применять определение нейтрализующей способности (титра) антител. Для анализа клеточно-опосредованного иммунного ответа допускается применять определение активности специфических CD4 и CD8 лимфоцитов (например, методом иммуноферментных пятен (Elispot)).
67. При исследовании на вирусную нагрузку ее допускается оценивать методом титрования в культуре клеток или количественного определения нуклеиновых кислот.
Вторичная фармакодинамика и фармакологическая безопасность
68. Оценка эффектов, оказываемых на респираторную и сердечно-сосудистую системы, может быть проведена в исследованиях на обезьянах, предпочтительно на животных, использовавшихся в исследованиях по первичной фармакодинамике. Эффекты на центральную нервную систему оценивают в исследованиях нейровирулентности, описание которых приведено пункте 70 настоящей главы.
5.3. Фармакокинетика
69. Исследования фармакокинетики неприменимы к данным типам вирусных вакцин.
5.4. Токсикологические исследования
Вирулентность
70. Считается, что вирулентность зависит от репликации вируса в месте введения и диффузии его в кровь. Необходимо представить подробное описание животной модели, используемой для оценки вирулентности, в отношении патогенеза и критериев приемлемости при оценке исходов. Местная репликация вируса может быть исследована на модели мышей при внутрикожном введении в ушную раковину, а также при оценке дозозависимой выживаемости после аэрозольного применения.
Нейровирулентность
71. Для вакцин против оспы не установлена модель для оценки нейровирулентности, которая была бы напрямую применима к возникновению поствакцинального энцефалита у человека.
72. Нейропатогенность вакцины против оспы определяют по способности вируса проникать через гематоэнцефалический барьер (нейроинвазивности) и по местной репликации вируса в головном мозге. Оба эффекта необходимо независимо исследовать на различных моделях.
73. Возможность проникновения через гематоэнцефалический барьер допускается исследовать на мышиной модели путем индукции виремии при интраназальном пути введения. Наличие вируса в головном мозге необходимо ассоциировать с маркерами энцефалита, такими как местное увеличение факторов некроза опухоли и интерлейкина-1 матричной РНК. Необходимо рассмотреть возможность непосредственного введения вируса в мозг (этмоидально), особенно в случае раннего энцефалита.
74. Способность вакцинного вируса реплицироваться непосредственно в головном мозге должна быть исследована путем непосредственного введения его в мозг (например, на неполовозрелых мышах).
75. Для обеих моделей необходимо предусмотреть гистологические исследования на предмет повреждения тканей, репликации вируса в тканях мозга и исследования иммуногенности на одних и тех же животных.
76. Для оценки и валидации результата исследования на нейровирулентность необходимо использовать положительный контрольный материал, такой как высоконейровирулентный штамм ортопоксвируса (например, штамм Western Reserve вируса осповакцины).
Влияние на репродуктивную функцию
77. Согласно научным медицинским данным, известно, что вакцинация у женщин в первом триместре беременности может спровоцировать выкидыш и пороки развития плода, в то время как в более поздние периоды беременности риск развития повреждений у плода не превышает такой риск для невакцинированных женщин. Токсическое влияние на репродуктивную функцию может быть вызвано эффектами, полученными в результате иммунного ответа на введение вакцины или связано с размножением вируса и проникновением его в утробный плод. Необходимо принять меры для того, чтобы вакцины против оспы не вводились женщинам, которые потенциально могут быть беременными.
78. В случае экстренных ситуаций может возникнуть необходимость введения вакцины против оспы на ранних сроках беременности. Так как риски вакцинации во время беременности недостаточно изучены, необходимо провести специальные исследования с целью определения возможного повышенного периода риска на ранних этапах беременности, что поможет ввести строгое противопоказание для вакцинации в указанный период. Репродуктивная токсичность может быть исследована на животных, например, мышах или кроликах, при внутрикожном введении вакцины-кандидата или вакцины-сравнения. Для должного дизайна исследования необходимо предусмотреть однократное введение вакцины определенным группам животных за несколько дней до или после спаривания. При этом необходимо предусмотреть введение дополнительных групп животных, которым однократное введение вакцины будет сделано в более поздние периоды времени на протяжении беременности.
Мутагенность и канцерогенность
79. Исследования по изучению мутагенности и канцерогенности неприменимы к данным типам вирусных вакцин.
Местная переносимость
80. Необходимо оценить местную переносимость конечного продукта. В качестве животной модели можно использовать, например кроликов. Местную токсичность наблюдают до образования оспин. В некоторых случаях возможные местные эффекты могут быть оценены в исследованиях токсичности при однократном и повторном (многократном) введении, таким образом устраняя необходимость в проведении отдельных исследований местной переносимости.
6. Клинические исследования
6.1. Основные положения программы клинической разработки
81. Клиническая оценка новой вакцины должна включать в себя: оценку иммунного ответа на основной антиген (антигены); исследования с целью оценки протективной эффективности; документирование профиля безопасности вакцины, включая информацию о местной реактогенности и ранних и отдаленных побочных эффектах.
82. Ввиду того, что вирус натуральной оспы в настоящее время не циркулирует в популяции, проведение исследований протективной эффективности невозможно. Следовательно, потенциальная протективная эффективность новой вакцины против оспы должна быть оценена, исходя из других параметров.
83. До глобального искоренения оспы формирование оспины должного размера с последующим образованием струпа и рубцеванием в месте первичной инокуляции коррелировало с профилактической эффективностью против инфекции. В частности, площадь поверхности рубца, также как и количество рубцов от предыдущих иммунизаций, находилось в обратной зависимости с летальностью. После успешной вакцинации, предполагаемая продолжительность протективного эффекта была не менее 3 лет, при этом частичная, в той или иной мере, протективность сохранялась на протяжении 10 и более лет.
84. Имеются исследовательские данные, что после первичной вакцинации, образование небольшого очага повреждения (оспины или язвы), диаметром 1 - 8 мм, было связано с максимальными уровнями нейтрализующих антител, хотя данные по методологии данных исследований не всегда доступны. Связь между размером повреждения и уровнями антител, подтвержденная в исследованиях с использованием реакции торможения агглютинации, представляется гораздо более слабой. Данные исследования применимы как к вакцинным штаммам NYCBOH (выращенным на лимфе телят или в куриных эмбрионах), так и к штаммам Lister/Elstree).
85. Исходя из положений пункта 84 данные по потенциальной протективной эффективности новой вакцины против оспы допускается получать основываясь на доле вакцинированных у которых образовались оспины необходимого размера в месте введения вакцины. Хотя предсказательная ценность лабораторной оценки гуморального и клеточного иммунного ответа у людей не доказана, предыдущие наблюдения и данные, полученные в ходе исследований на животных, доказывают необходимость проведения подобных лабораторных тестов. Следует исследовать связь между результатами данных тестов и формированием оспин.
86. Профиль безопасности противооспенных вакцин, использовавшихся до прекращения рутинной вакцинации, которое произошло после глобальной эрадикации заболевания, подробно описан. Серьезные и угрожающие жизни побочные реакции случались редко или очень редко. Тем не менее текущее отсутствие циркулирующего в популяции вируса натуральной оспы может влиять на соотношение «польза - риск» в отношении вакцинации. Более того, в настоящее время вакцинация проводится в основном только в отношении персонала исследовательских центров, где проводится работа с ортопоксвирусами.
87. Количество субъектов, подвергшихся воздействию новой противооспенной вакцины в клинических исследованиях, должно поддерживаться на минимальном уровне, но необходимом для обеспечения в достаточной мере оценки ее потенциальной протективной активности и безопасности.
6.2. Оценка иммунного ответа и конечные точки
Наличие и размеры кожной реакции (оспины)
88. Учитывая предшествующий опыт использования вакцин, новая противооспенная вакцина должна индуцировать образование характерных кожных оспенных реакций у, по крайней мере, 95% здоровых реципиентов после первичной иммунизации. Определение образования оспины должно основываться на внешнем виде эриматозной папулы или пустулы в месте введения в течение одной недели после вакцинации.
89. Необходимо, чтобы все очаги повреждения были подробно описаны в протоколе исследования по внешнему виду, размеру (в сравнении с градуированной шкалой) и времени первого появления. Необходимо зафиксировать время, прошедшее до начала коркообразования (которое позволяет оценить общую продолжительность вирусовыделения) и до отпадения корок. Хотя добровольцы могут самостоятельно записывать большую часть данной информации в дневниках пациентов, необходимо запланировать включение в исследование количество визитов, достаточное для визуальной оценки указанных процессов исследователями. Необходимо осуществлять рутинную фотографическую фиксацию поражений.
Иммунный ответ
90. Гуморальный и клеточный иммунный ответ, должны быть охарактеризованы по результатам клинического изучения вакцины против оспы. Также должна быть исследована связь между результатами проведенных исследований и формированием оспин. Необходимо предпринять меры для подтверждения связи между иммунологическими параметрами и образованием оспин.
91. Оценка гуморального иммунного ответа должна включать обнаружение и определение титра нейтрализующих антител с использованием внутриклеточного зрелого вириона (intracellular mature virion, IMV) в отношении соответствующего рабочего (вторичного) стандартного образца, откалиброванного по подходящему первичному стандартному образцу. При использовании любых новых технологий (включая методики иммуноферментного анализа) они должны быть валидированы в отношении исследований по нейтрализации антител и должны включать дифференцированную оценку IgG и IgM ответов.
92. Исследования по оценке клеточного иммунного ответа должны включать оценку активности CD8 Т-клеток с применением чувствительных методик, таких как активация клеточного иммунитета с использованием живого вируса и продукции интерферона гамма (например, методами Elispot и проточной цитометрии).
6.3. Дизайн клинических исследований
Фармакологические исследования
93. Неконтролируемые клинические исследования в небольших группах здоровых взрослых добровольцев должны быть достаточными для предварительного описания профиля безопасности и иммуногенности новой вакцины. Тем не менее, в зависимости от результатов доклинических исследований, на данном этапе может возникнуть необходимость в проведении сравнительных исследований с использованием вакцин, содержащих различное количество бляшкообразующих единиц вируса осповакцины в 1 мл (PFU/ml).
94. Исследования должны проводиться на добровольцах, ранее не вакцинированных против оспы. В случае если имеются сомнения относительно того, был ли подвержен доброволец воздействию вируса осповакцины или других ортопоксвирусов, для подтверждения его соответствия указанному критерию отбора, необходимо до начала иммунизации провести высокочувствительные иммунологические тесты по обнаружению CD4 Т-лимфоцитов (например, исследования на лимфопролиферацию). Необходимо предпринять все возможные усилия, чтобы исключить из исследования субъектов, имеющих повышенный риск развития побочных реакций на введение вакцины, содержащей живой аттенуированный вирус осповакцины. Особое внимание необходимо уделить исключению из исследования субъектов, имеющих в медицинской истории случаи любого атопического (не только экзема) дерматита и (или) страдающих на момент исследования от активного кожного заболевания.
95. Данные исследования должны иметь необходимый масштаб, который позволит отразить потенциальный процент вакцинированных, у которых будут формироваться оспины. Любые возникшие повреждения кожи должны быть полностью описаны. Также необходимо получить данные относительно иммунного ответа со сбором образцов примерно на 4 - 6 неделе после вакцинирования. Необходимо обосновать количество добровольцев, участвующих в данных исследованиях, а также время проведения оценки иммунологических параметров.
Подтверждающие исследования иммуногенности
96. Данные исследования должны оценить воздействие новой вакцины на больших группах субъектов. При этом критерии отбора субъектов исследования должны быть подобны критериям отбора, применявшимся при ранних клинических исследованиях. В качестве подходящих субъектов не допускается включать в данные исследования детей и пожилых добровольцев. Из исследования необходимо исключить любых лиц с установленным риском развития побочных реакций на введение живого аттенуированного вируса осповакцины.
97. Если в результате предварительных исследований с участием человека будет установлено, что вакцины, содержащие различное количество бляшкообразующих единиц вируса осповакцины в 1 мл (PFU/ml), необходимо исследовать на большем количестве субъектов, то следует иметь в виду, что данные исследования должны быть двойными слепыми с рандомизацией согласно вводимой дозе PFU.
98. Необходимо провести рандомизированное двойное слепое исследование с целью продемонстрировать не меньшую эффективность новой вакцины по сравнению с зарегистрированной вакциной. Необходимо обосновать выбор границы не меньшей эффективности (дельта). В целях получения наиболее полных данных по безопасности нового продукта допускается использовать несбалансированную рандомизацию, при которой большинство субъектов исследования подвергаются воздействию незарегистрированной новой вакцины. Если доступно более одной подходящей для сравнения вакцины, необходимо провести сравнительные исследования только с одной из них, с учетом того, что выбор вакцины сравнения является обоснованным (при этом учитываются такие факторы, как тип вирусного штамма и доза).
99. Во всех описанных в пунктах 81 - 98 настоящей главы видах исследований необходимо провести оценку данных по лабораторному изучению различных типов иммунного ответа, по крайней мере у части субъектов исследования, в зависимости от общего количества добровольцев, подвергшихся воздействию.
Клинические исследования при отсутствии вакцины сравнения на таможенной территории Союза и за ее пределами
100. Применение вакцин, которые не соответствуют требованиям к разработке, изучению и производству биологических лекарственных препаратов допускается только в условиях чрезвычайных ситуаций и при отсутствии зарегистрированных на таможенной территории Союза или за ее пределами вакцин, разработанных и произведенных должным образом.
Исследование разрабатываемой (целевой) вакцины с вакциной сравнения не является обязательным при выполнении всех следующих условий:
отсутствие зарегистрированных на таможенной территории Союза или за ее пределами вакцин, разработанных и произведенных должным образом;
положительная оценка соотношения «польза-риск» для разрабатываемой (целевой) вакцины;
отсутствие циркулирующего в популяции вируса осповакцины.
101. Как указано в пунктах 88 и 89 настоящей главы, исходя из предшествующего опыта, априорно полагают, что вакцина должна вызывать образование отличительной оспины в месте введения у 95% вакцинированных. В условиях неконтролируемого клинического исследования необходимо рассчитать точность оценочного процента вакцинированных, у которых должны развиться оспины. При определении приблизительного количества субъектов, которых предполагается включить в исследование, необходимо учитывать данные доклинических исследований и ожидаемый процент вакцинированных (исходя из результатов предыдущих поисковых исследований), у которых разовьются оспины.
Клинические исследования при наличии вакцины сравнения на таможенной территории Союза и (или) за ее пределами
102. Если имеется доступная вакцина и (или) вакцина, зарегистрированная в Союзе до начала программы клинической разработки новой вакцины, или на ранних ее этапах, необходимо представить соответствующие данные сравнительных исследований. Выбор соответствующей вакцины сравнения для такого исследования должен быть одобрен уполномоченными органами (экспертными организациями) государств-членов.
Продолжительность приобретенного иммунитета
103. В настоящее время недостаточно установлена необходимость ревакцинации и оптимальное после первичной вакцинации время ее проведения. При этом имеющиеся рекомендации для ранее использовавшихся вакцин не всегда применимы к новым вакцинам.
104. При первичной регистрации новой вакцины заявление на ее регистрацию допускается подать задолго до того, как истечет один год с момента воздействия исследуемой вакцины на большинство субъектов исследования. В связи с этим в протоколах подтверждающих исследований по иммуногенности необходимо запланировать проведение повторных лабораторных исследований по оценке иммунного ответа через значительно более продолжительный промежуток времени, по крайней мере, в когорте субъектов исследования. Однако, в связи с недостаточностью информации в настоящее время, не предполагается, что в указанных протоколах будет запланировано введение последующих доз вакцины.
105. Детальные программы проведения оценки иммунного статуса должны быть представлены в составе регистрационного досье при первичной регистрации вакцины. Выполнение данных планов является частью пострегистрационных обязательств заявителя.
6.4. Оценка безопасности
106. Профиль безопасности нескольких вакцин против оспы первого поколения хорошо изучен. В научной медицинской литературе подробно описаны выявленные факторы риска развития различных типов нежелательных реакций. В зависимости от общего количества субъектов, включенных исследования иммуногенности, в целях оценки безопасности необходимо большую их часть включить в группу субъектов для оценки безопасности исследуемой вакцины. Для определения количества указанных субъектов исследования производитель вакцины должен учитывать исторические (анамнестические) данные по зарегистрированной частоте нежелательных реакций.
107. Поскольку количество субъектов, включенных в предрегистрационные исследования, не всегда достаточно для возможности выявления редких и очень редких побочных явлений, таких как энцефалит, объем полученных данных по безопасности должен быть существенным по крайней мере для возможности оценки частоты редких реакций. Необходимо оценить характеристики формирования оспин у вакцинированных с данными реакциями, при этом проведение исследований по иммуногенности не является обязательным. На момент первичной подачи заявления на регистрацию продолжительность последующего наблюдения с целью оценки безопасности должна быть не менее 3 месяцев для всех субъектов исследования, подвергшихся воздействию исследуемой вакцины. Соблюдение указанного периода необходимо для выявления возможного позднего развития нейротоксичности и случаев прогрессирующей вакцинии.
108. Необходимо учитывать, что даже известные осложнения вакцинации против оспы (как связанные напрямую с репликацией аттенуированного вируса, так и несвязанные) не всегда полностью спрогнозированы и предотвращены путем тщательного анализа историй болезни вакцинируемых. В настоящее время не имеется валидированных методов лечения вакцинно-ассоциированных осложнений. Отсутствие таких методов является главной причиной существующих указаний, по которым общее количество субъектов клинических исследований, должно оставаться на минимально необходимом уровне. Вместе с тем в протоколы исследований необходимо включить информацию о любых возможных мерах неотложной медицинской помощи при подобных осложнениях, исходя из актуальной информации о частоте и течении подобных осложнений и доступности потенциально эффективных лекарственных препаратов. Во всех случаях применения специфического лечения для подобных осложнений необходимо тщательно зафиксировать принятые терапевтические мероприятия, проследить и задокументировать клинические исходы.
109. Дополнительно в случае продолжительной или тяжелой лихорадки после вакцинации (являющейся вероятным признаком виремии), необходимо установить наличие виремии, используя признанные и (или) экспериментальные вирусологические методы. За пациентами с указанными симптомами необходимо установить тщательное последующее наблюдение.
6.5. Пострегистрационные исследования
110. В связи с особенностями разработки вакцин против оспы второго поколения их первичная регистрация на условиях продолжается (в исключительных случаях) вплоть до положительного завершения всех продолжающихся исследований в ходе выполнения заявителем своих обязательств по пострегистрационному мониторингу.
111. Поскольку не ожидается возникновения вакцинноассоциированных побочных явлений позднее 3 месяцев после вакцинации, необходимо проводить активный мониторинг вакцинированных на протяжении большего периода времени, продолжительность которого должна быть обоснована. В протоколе исследования необходимо запланировать длительное последующее наблюдение за разными видами иммунного ответа (по крайней мере в когорте вакцинированных).
112. В зависимости от исследований, проведенных в когортах вакцинированных, за которыми проводилось длительное последующее наблюдение в отношении иммунного ответа, необходимо провести сравнение этих когорт вакцинированных с когортой добровольцев, не имевших антител к вирусу осповакцины. Сравнение производится по проценту вакцинированных, у которых сформировались оспины и по формированию разных видов иммунного ответа у вакцинированных.
113. Если этого не было сделано ранее в зависимости от результатов исследований у здоровых добровольцев, на данной стадии обосновано проведение исследований безопасности и иммуногенности у здоровых детей и пожилых.
114. В случае чрезвычайной ситуации может потребоваться вакцинация лиц, которых при обычных обстоятельствах, не иммунизируют живым вирусом осповакцины (например, беременных женщин, лиц с иммунодефицитом, страдающих аллергическими заболеваниями). Протоколы исследований должны быть разработаны таким образом, чтобы в данной специфической ситуации были собраны все важные данные, касающиеся формирования оспин, иммуногенности и безопасности у этих субъектов. Учитывая обстоятельства, в которых будет осуществляться сбор этих данных, заявителям необходимо разрабатывать такие протоколы совместно с органами системы здравоохранения государств-членов.».
Обзор документа
Скорректированы правила проведения исследований биологических лекарственных средств ЕАЭС.
Урегулированы исследования препаратов из плазмы крови, препаратов на основе терапевтических белков, вакцин для профилактики гриппа и оспы. Оговорено снижение риска передачи возбудителей прионовых инфекций с этими группами препаратов.
Решение вступает в силу по истечении 180 дней с даты опубликования.